

Summary:
The Investigational New Drug (IND) process is a regulatory pathway followed by pharmaceutical companies and researchers to obtain permission from regulatory authorities, such as the U.S. Food and Drug Administration (FDA), to conduct clinical trials with a new drug or biologic in humans. The Food and Drug Administration is responsible for regulating all pharmaceutical drugs that are available on the USA’s market, but at what point do they become involved? A Investigational New Drug (IND) Application introduces a new drug into the drug regulatory system, and is necessary before a drug can be administered as part of a clinical trial.
The first step of completing an IND is the pre-IND meeting, which drug manufacturers should be prepared to have towards the end of the preclinical development process. But what does an IND involve? In this article we will discuss the three aspects needed for an IND application, and how alternative animal models can help lower the cost and timeline associated with clinical drug development.
Drug discovery is a complex process that includes several stages, such as target identification, lead identification, lead optimization, preclinical development, and clinical trials [Figure 1]. On average, bringing these drugs to market takes between seven to ten years and requires an investment of approximately one billion dollars per drug.
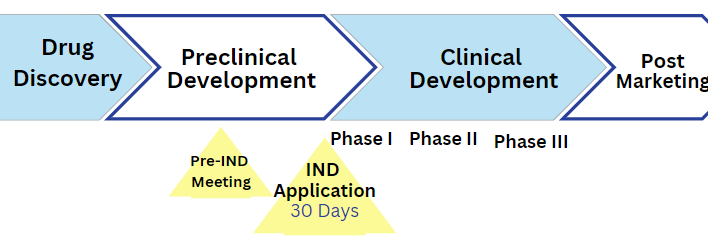
Figure 1. New Drug Development and Review Process
Effective communication between drug manufacturers and the FDA, which oversees drug regulations in the USA, is a critical aspect of the drug discovery process. The initial point of contact between the two parties is the Investigational New Drug (IND) Application which the manufacturer or potential marketer submits when they have a drug compound that is ready to enter clinical trials (U.S. FDA, 2022). Manufacturers also have the option to request a pre-IND meeting with FDA staff in order to discuss the manufacturer’s IND application.
Pre-IND Meeting
This pre-IND meeting is valuable as it lets the manufacturer gain feedback and guidance on their IND application which can help avoid an application being considered deficient, and being put on clinical hold. Beyond ensuring that the IND application has the necessary components, the pre-IND meeting can shorten the approval time, as the FDA staff is familiar with the research and the proposed study. But what is needed for this pre-IND meeting, and the IND application itself?
It is important to enter into the pre-IND meeting with:
- Chemistry, Manufacturing, and Control (CMC) Information
- Preclinical Support
- Detailed Clinical trial design
- Showcase compliance with Good Clinical Practices (GCPs)
- Selection of Dosage information
Based on this pre-IND discussion, the FDA can recommend ways to bring the drug to market faster, such as advising on whether a drug qualifies for an accelerated track designation.
What Qualifies as Good Preclinical Data to Include in your Pre-IND Meeting and IND Application?
One of the most commonly asked questions about the IND application is what qualifies as good preclinical support, as manufacturers often find themselves toeing a line between trying to lower the notoriously high cost of drug development, while obtaining sufficient quality data to showcase the pharmacological benefit of their compounds. This is where alternative animal models such as zebrafish and C. elegans can be hugely advantageous, as they offer whole organisms testing that is fast and cost effective.
While, traditionally, drug development has been predominantly conducted on rodent models, recently, the FDA has signaled a desire to adopt a ‘fail fast, fail safe’ approach, that promotes the utilization of alternative models. For instance, the recently passed Modernization Act made it so that animal testing was no longer required in the preclinical stage of drug development. While speaking in support of the law, Senator Rand Paul explained the goal of the new legislation, saying that it aimed to “accelerate innovation and get safer, more effective drugs to market more quickly by cutting red tape that is not supported by current science (Paul, 2023).”
Thus, if manufacturers are able to present a data package during their pre-IND meeting that clearly shows improvement, even if it is done in an alternative animal model, such as a zebrafish or C. elegans, then they’re in a very good position.
The FDA doesn’t require any particular model system, mouse models are just the de facto standard, but this is to our industry’s detriment, because not only are rodents expensive, they aren’t always the best way to model.
President of Vivifica Pharmaceuticals
How Alternative Animal Models Can Help the Drug Discovery Process:
Here at InVivo Biosystems, we partner with pharmaceutical and biotech companies to generate the comprehensive science-based evidence of safety and efficacy that is needed for the IND application. One of our current pharmaceutical clients explained why they chose to reach out to us, and what they see as the benefit to employing alternative animal models to the drug development process – saying that companies such as theirs face both internal company pressure and external public pressure to transition towards a way of testing that is more effective. After all, currently, only around 10% of drugs that enter human clinical trials ultimately receive approval, which goes to show that the current system is not working (Sun, Gao, Hu & Zhou, 2022).
Working with InVivo Biosystems gave us the information [we needed] to better improve our and optimize our compounds to the final clinical candidate stage
Dr Marius Galyan, Seventeen Minutes of Science, Episode 66
Beyond having the advantages of lower cost and accelerating a project’s timeline, alternative animal models may help improve the current high drug failure rate. This is because, alternative animal models can help to identify potential adverse effects of a drug candidate which may not be evident in traditional mammalian models, for instance, some drugs induce immunological reactions and toxic effects on humans’ cardiovascular systems which are not observable in rodents (Lee, Hwang & Kim, 2017). Likewise, neurological diseases such as Alzheimer’s and Parkinson’s are not well modeled in rodents, as, mouse models do not fully recapitulate the pathological features seen in Alzheimer’s disease, and the current Parkinson’s models are limited in their inability to reproduce the selective vulnerability of dopaminergic neurons that is seen in humans (Dawson & Ko, 2013).

Conclusion:
The IND application is an essential requirement for drug manufacturers seeking to enter the pharmaceutical market in the USA by conducting clinical trials. Recent technological advancements have motivated researchers to explore alternative animal models for studying diseases traditionally studied in rodents. These novel models have the potential to revolutionize drug development, as they not only exhibit closer resemblance to human diseases in certain cases but also offer a faster and cost-effective means to generate the comprehensive data package necessary for an IND application.
References:
- U.S. Food and Drug Administration (2/22/22). Small Business and Industry Assistance: Frequently Asked Questions – Pre-Investigational New Drug (IND). Retrieved May 16, 2023, from https://www.fda.gov/drugs/cder-small-business-industry-assistance-sbia/small-business-and-industry-assistance-frequently-asked-questions-pre-investigational-new-drug-ind
- U.S. Food and Drug Administration. (2/28/22). Investigational New Drug (IND) Application. https://www.fda.gov/drugs/types-applications/investigational-new-drug-ind-application
- Paul, S. (2023). Dr. Paul’s Bipartisan FDA Modernization Act 2.0 to End Animal Testing Mandates Included in 2022 Year-end Legislation, Dr Rand Paul, US Senator, Kentucky. https://www.paul.senate.gov/news/dr-pauls-bipartisan-fda-modernization-act-20-end-animal-testing-mandates-included-2022-year
- Dawson, T. M., & Ko, H. S. (2013). Alzheimer’s disease: emerging concepts and therapeutic targets. Journal of neurochemistry, 127(3), 454-461. doi: 10.1111/jnc.12194
- Lee, E. J., Hwang, Y. H., & Kim, H. R. (2017). Non-clinical safety evaluation of therapeutic monoclonal antibodies: current status and future directions. Journal of pharmaceutical investigation, 47(4), 287-296. doi: 10.1007/s40005-017-0323-x.
- Sun, D., Gao, W., Hu, H., & Zhou, S. (2022). Why 90% of clinical drug development fails and how to improve it?. Acta pharmaceutica Sinica. B, 12(7), 3049–3062. https://doi.org/10.1016/j.apsb.2022.02.002

