

Summary
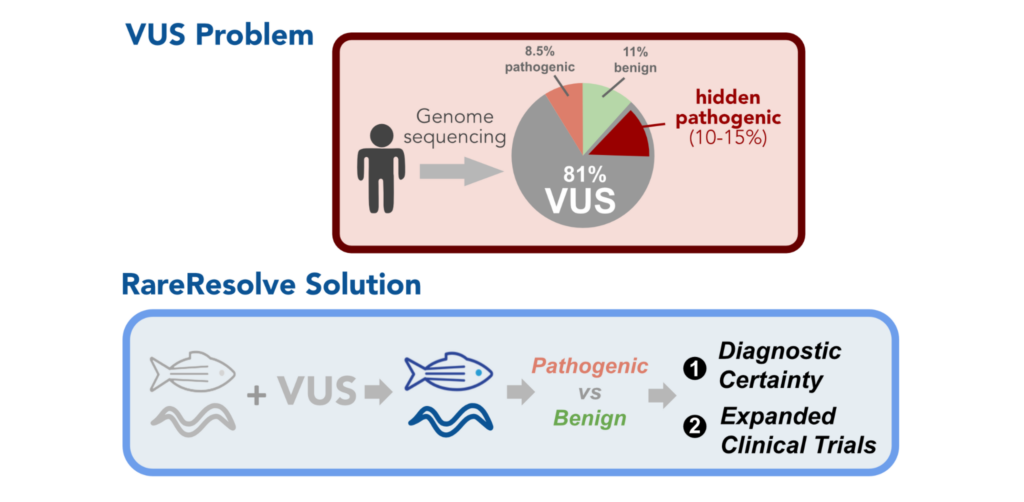
Of the over 60 million people worldwide who suffer from epilepsy, approximately one third of them do not respond to available therapies. While genetic testing has revealed underlying genetic variants of the disease, it has also expanded the list of Variants of Uncertain Significance (VUS), an enigmatic designation which sows uncertainty into diagnosis and clinical decision making. Variations in the STXBP1 gene are linked with several forms of epilepsy and over 100 distinct variants in STXBP1 have been reported. The InVivo Biosystems team are experts in modeling and classifying both known variants and VUS, particularly with STXBP1. Our research team is intimately familiar with the importance of individual variant contributions to patient disease presentation and progression as well as treatment. In Part I of this blog, we described how we modeled 80 VUS in STXBP1 using humanized C. elegans combined with deep phenotyping and machine learning. To complement the expansive body of work undertaken to model STXBP1 variant pathogenicity in C. elegans,2,3 the team has also evaluated the use of zebrafish to model multiple patient mutations in a well-conserved ortholog of human STXBP1. In this blog post, we’ll explore the creation of these zebrafish models and their utility in the functional validation of VUS, shedding light on their importance for both rare disease research and drug screening efforts.Strengths of Non-Mammalian Models
Traditionally, mammalian models like mice have been instrumental in genetic research due to their genetic and physiological similarities to humans. However, for VUS functional validation, they have limitations. Mammalian models are costly, time-consuming, and ethically sensitive. Additionally, their complex genetic makeup can sometimes obscure the specific effects of a single genetic alteration. Non-mammalian organisms, such as zebrafish (Danio rerio), fruit flies (Drosophila melanogaster), and nematode worms (Caenorhabditis elegans), have become invaluable assets in the study of VUS.4 These organisms share a surprising amount of genetic and biological conservation with humans. Their simple genetic makeup, short lifespans, and ease of genetic manipulation make them ideal candidates for functional validation studies.- Simplicity: Reduced complexity makes it easier to isolate and study the effects of specific genetic alterations.
- High Throughput Screening: Enable researchers to study multiple genetic variants simultaneously, accelerating the research process.
- Genetic Manipulation: Techniques like CRISPR-Cas9 gene editing enable precise modification of the genome, facilitating the study of specific genetic variants.
- Cost-Effectiveness: Maintenance of non-mammalian organisms is considerably more affordable than working with mammals, allowing for larger sample sizes and broader studies.
Modeling in Zebrafish (Danio rerio)
Similar to C. elegans, zebrafish have been increasingly used in recent years as non-mammalian alternatives to modeling epilepsy, with both pharmacological and genetic epilepsy models being widely used in drug discovery research to date.4,5 Their external development, transparency during early development, relatively compact housing, small embryonic & larval size, and large clutch sizes have facilitated numerous preclinical high throughput library screens for antiepileptic compounds. Impressively, one such drug repurposing study in a genetic model of Dravet Syndrome led to discovery and a compassionate use clinical trial of a compound found to ameliorate seizure behaviors,6,7 highlighting the potential for zebrafish to accelerate such drug discovery efforts for many other genetic disorders.
Our approach to building an epilepsy model by targeting STXBP1 was prompted in part by work in the Baraban lab showing that mutations in the zebrafish paralogs stxbp1a or stxpb1b produced phenotypes that emulated epileptic seizure activity.8 Both stxbp1a and stxbp1b have high pairwise sequence identity (87% and 79%) to human STXBP1. Of the two genes, we reasoned that stxbp1a would be more favorable for modeling due to a more robust phenotype and broader expression pattern. As we have noted from our experience generating C. elegans whole gene humanized animal models, having a consistent – and ideally strong – loss of function (LOF) phenotype is an important feature for genetic models intended for compound library screening. This helps ensure that effects of compounds that ameliorate LOF phenotypes are readily apparent and not lost in phenotypic “noise” from subtler, more variable LOF phenotypes.
CRISPR Model Creation
To generate patient-specific zebrafish models of STXBP1-related disorders, the team initially selected two patient-derived variants—one predicted as pathogenic and another predicted as benign—to be constructed in parallel. Humanization by whole gene replacement was and continues to be a challenge in the field. Therefore, the team opted to capitalize on the high level of conservation at the amino acid level by selecting variants with conserved residues in the orthologous zebrafish protein. The team hypothesized that the predicted pathogenic variant (stxbp1a Ser42Pro) would exhibit a phenotype similar to the loss of function (LOF), while the benign mutant (stxbp1a Pro94Leu) would closely resemble the wild type.
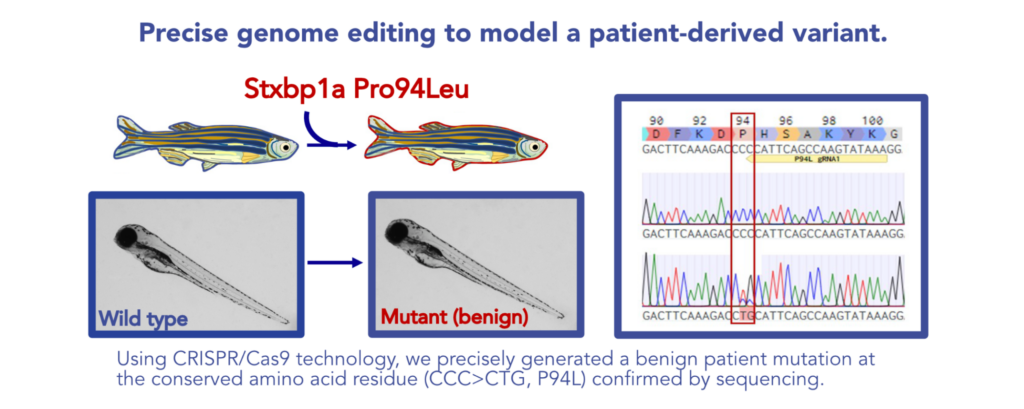
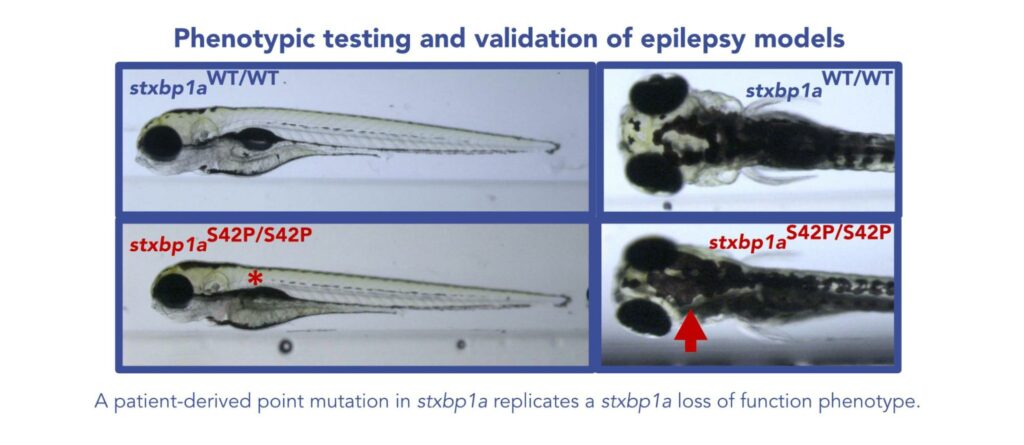
To introduce these patient variants into the zebrafish genome, the team deployed our CRISPR/Cas9-mediated knock-in (KI) technology to precisely edit the selected sites within a clean genetic background. Thorough sequencing following outcrossing and selection for homozygotes confirmed the accuracy of the edits in the two new mutant fish lines. At initial assessment, the pathogenic Ser42Pro mutant exhibited the distinct stxbp1a hyperpigmentation phenotype reported previously, whereas the benign Pro94Leu mutant did not. This suggested that the Ser42Pro mutation was impacting some aspect of stxbp1a function. However, the critical test would be whether it captured a disease phenotype in a way that would bolster the validity of variant classification and potentially serve as a platform for therapeutic screening.
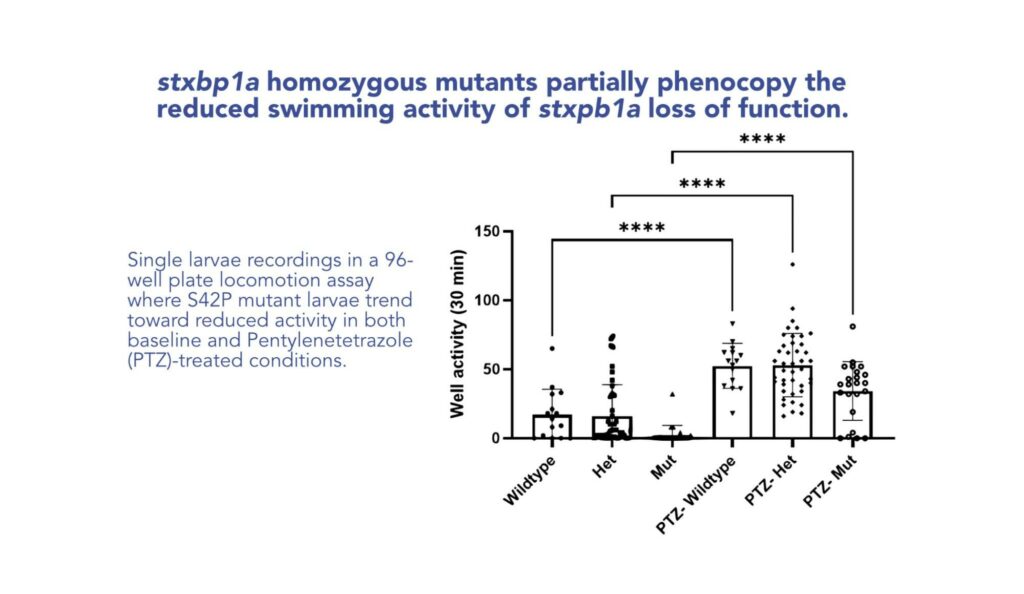
To test for a neurological phenotype, the pathogenic and benign variants were tested in a high-throughput activity assay. In these assays, the baseline activity of the S42P mutant indicated an almost complete loss of activity, similarly to the stxbp1a knock-out (KO). This stark and consistent phenotype provides a promising basis for variant classification. In contrast with the stxbp1a KO, however, the S42P mutant retained some gain of activity in response to pentylenetetrazol (PTZ), a compound commonly used to model epileptic seizures in mice and zebrafish. Consistent with the model, the line bearing the benign P94L mutation showed no detectable loss of swimming activity.
KO vs. KI: Loss-of-Function Knock-out or Variant Knock-In?
Why make the effort to construct a variant KI model when a KO line exists with a relevant LOF phenotype? The phenotypic differences shown in this case study illustrate why. With the variant knock-in, the model can more accurately capture the variant-specific contributions to function and differences in clinical presentation. The complete loss of the gene and protein product inherently removes the leading drug target from subsequent drug library screens. Also, among the well-characterized pathogenic mutations detected in the clinic, many are not homozygous recessive but rather autosomal dominant heterozygous carriers or other types of alleles where a full KO is not an accurate model. Furthermore, the absence of the gene could trigger compensatory gene expression that would confound the interpretation of results. Owing to the robust but differential phenotype of the S42P KI mutant relative to the KO, we are now in the design phase of a compound library screen leveraging the S42P mutant.
Tools to resolve uncertainty in variant classification
This proof-of-principle with a single predicted pathogenic stxbp1a variant instills great promise for improving variant classification and therapeutic testing. In C.elegans we successfully analyzed 80 patient-derived humanized variants and used the model to generate hypothetical classifications of pathogenic or benign designations, which substantially increased the diagnostic yield.2 These stories represent just two of the tools we have for confronting the expanding uncertainty arising from VUS and illustrate how evidence from multiple model systems can synergistically improve diagnoses. Whether you are looking to identify and validate a target for modeling, or aiming to tailor a model for a drug screening platform, the team at InVivo Biosystems offers a range of services to empower researchers at early stages of development.
Humanized Models for Therapeutic Discovery with RareResolve™
RareResolve™ introduces an innovative and precise animal modeling platform, utilizing genetically-engineered zebrafish with human gene expression through our Whole-gene Humanized Animal Model (WHAM) method. By substituting the animal’s gene with the human coding sequence for the target disease gene, this approach minimizes false outcomes in drug testing. The WHAM-humanized platform utilizes expression of human genes with or without pathogenic variants, facilitating more accurate diagnosis of VUS and validation of candidate medicines. RareResolve™ builds upon this method to accelerate drug discovery by integrating two key capabilities:
1) Rapid gene knockouts in F0 animals, known as “crispants”.
2) Robust human transgene expression from injected mRNA.
By combining these methods into a rapid and cost-effective platform, RareResolve™ allows medium-throughput screening of hundreds of hits, making it an ideal approach in the rare disease space. This innovative system aligns with the evolving needs of personalized medicine, offering a fast, accurate, and affordable solution for preclinical research.
Fast Target Validation with RapidGen™
Our novel RapidGen™ model enables creation and validation of a large number of alleles within one month! InVivo Biosystems optimized the crispant approach9 to efficiently “break” a gene at multiple sites using multiple sgRNAs. The result is a highly penetrant gene knockout that can be assayed by phenotype immediately in the injected F0 generation animals. A traditional CRISPR-based edit modifies a single site in a gene and requires months of propagation, crossing, and screening to stabilize and confirm the edit before assaying. Because the crispant approach enables immediate screening in the injected F0 animals, large numbers of targets can be screened in a high-throughput format. RapidGen™ enables researchers to keep up with the fast pace of discovery by rapidly validating multiple targets and optimizing a model before committing time and resources to therapeutic testing.
RapidGen Fish Model | InVivo Biosystems
Conclusions
At InVivo Biosystems, one of our goals is to leverage the speed, economy, and efficacy of small animal models to accelerate the discovery of novel therapeutics. Non-mammalian model organisms have been pivotal in understanding various rare diseases, including certain types of muscular dystrophy, neurodegenerative disorders, and rare genetic syndromes. Our expertise and capability of quickly creating these economical models have led to several partnerships with rare disease organizations. By introducing VUS into these models, researchers can observe the phenotypic changes and gain insights into the variant’s functional implications. Understanding the functional consequences of VUS is not only crucial for rare disease research but also for drug screening efforts. Non-mammalian models can be used to test potential therapies and drug compounds swiftly. By introducing VUS into these models, researchers can assess the efficacy of drugs, leading to the development of targeted treatments in an accelerated manner compared to traditional mammalian models. This can greatly accelerate therapeutic development pipelines, making a meaningful impact on the lives of those impacted by rare diseases and disorders.
If interested in learning more about our revolutionary technology, request a consultation with one of our experts below:
References
- Remy, S. & Beck, H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 129, 18–35 (2006).
- Hopkins, C. E. et al. Clinical variants in Caenorhabditis elegans expressing human STXBP1 reveal a novel class of pathogenic variants and classify variants of uncertain significance. Genet. Med. Open 1, 100823 (2023).
- Zhu, B. et al. Functional analysis of epilepsy‐associated variants in STXBP1/Munc18‐1 using humanized Caenorhabditis elegans. Epilepsia 61, 810–821 (2020).
- Johan Arief, M. F., Choo, B. K. M., Yap, J. L., Kumari, Y. & Shaikh, M. F. A Systematic Review on Non-mammalian Models in Epilepsy Research. Front. Pharmacol. 9, 655 (2018).
- D’Amora, M. et al. Zebrafish as an innovative tool for epilepsy modeling: state of the art and potential future directions. Int. J. Mol. Sci. 24, 7702 (2023).
- Baraban, S. C., Dinday, M. T. & Hortopan, G. A. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet Syndrome treatment. Nat. Commun. 4, 2410 (2013).
- Griffin, A. et al. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain aww342 (2017) doi:10.1093/brain/aww342.
- Grone, B. P., Qu, T. & Baraban, S. C. Behavioral Comorbidities and Drug Treatments in a Zebrafish scn1lab Model of Dravet Syndrome. eneuro 4, ENEURO.0066-17.2017 (2017).
- Kroll, F. et al. A simple and effective f0 knockout method for rapid screening of behaviour and other complex phenotypes. eLife 10, 1–34 (2021).


