

Summary:
Mutations in the STXBP1 gene are implicated in childhood epilepsies and several neurodevelopmental disorders, however, nearly half of the STXBP1 variants recorded in the ClinVar database are Variants of Unknown Significance (VUS). VUS are a notable burden on our healthcare industry – extending the timeline for patients to receive adequate treatment, and impending the development of new, targeted, drug therapies. Here at InVivo Biosystems we have leveraged our expertise in CRISPR to develop humanized C. elegans and zebrafish models of STXBP1. This article will briefly expand upon STXBP1 before diving into our recent work, discussing these humanized platforms which can enable the classification of variants as benign or pathogenic, and the implications these model organisms have on rare disease research.
STXBP1 is a protein involved in synaptic vesicle trafficking. Pathogenic variants (mutations) in the STXBP1 gene cause a variety of disorders including: childhood epilepsy, Global Delay, Cognitive impairment (mild to profound), movement disorders, and Autism (STXBP1 Foundation, 2021). While there are symptoms (delays in developmental milestones and early onset seizures) which are suggestive of a STXBP1-related disorder, genetic testing is required to reach a diagnosis (CHOP, 2021). However, diagnosis of STXBP1-related epilepsies, and other genetic disorders, can be impeded by Variants of Unknown Significance (VUS). These are variants (mutations) which have an unknown effect on disease, and thus, when they are found in patients present a conundrum. In a recent blog post, we examined the increase in VUS as seen through the lens of in silico calculations, further highlighting the need for rapid classification. In a most recent update of the data from ClinVar as of October 2023, there are now 1,136,575 million missense variants in the ClinVar data-base and 926,376 of these variants are classified as VUS (82% of all missense data). [Figure 1] In the two years since our prior post, the number of VUS have more than doubled in the database (up 230%), while the number of variants that have been categorized has grown by less than 40% (Hopkins et al., 2023). If the data continues to grow in this way, in the future, 98% of the variants in the ClinVar database will be unactionable.
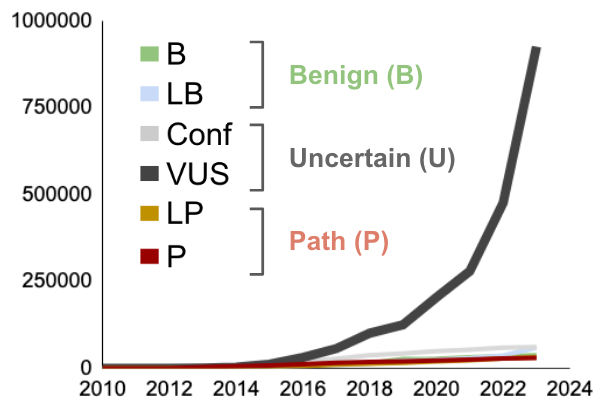
Figure 1. Growing need for classification in reported variants of STXBP1. A. Number of missense variants in ClinVar that are VUS (dark gray), pathogenic (red), likely pathogenic (pink), likely benign (light green), or benign (green) as a function of time (Hopkins et al., 2023).
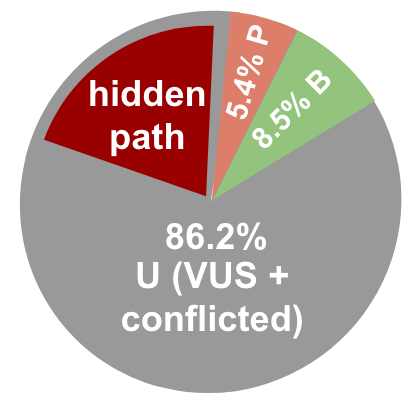
This data clearly shows that something needs to be done now to actively begin classifying VUS variants. Recognizing this need, and how critical diagnosis is for individuals to receive the medical care and treatment they need, the National Human Genome Research Institute has published a set of ‘strategic visions’ which aim to render the term “VUS” obsolete by 2030 (Green et al., 2020). However there is a large fraction of VUS that are hiding pathogenic alleles.
Commonly used methods for variant classification include in silico tools and functional characterization in animal models such as rodent models and other model organisms such as C. elegans and zebrafish. While in silico methods can provide insights and offer a fast, inexpensive way to assess thousands of variants at once, they come with their own unique set of limitations; most notably, they are only moderately sensitive tests and need functional data to support their predictions (Gunning et al., 2021). Similarly, while rodent models have a high degree of genetic conservation in human disease-associated genes and provide accurate functional data, they are challenging and costly to generate.
For these reasons, when wanting to address the need for better, faster, variant classification, members of the InVivo Biosystems team decided to generate humanized C. elegans (using gene replacement) and zebrafish (using patient specific point mutations) models of STXBP1 dysfunction utilizing CRISPR/Cas9-mediated genome editing. These models enabled the team to classify variants as benign or pathogenic, and demonstrated how advantageous these alternative model organisms can be for epilepsy research when compared to mice (the most commonly used model for STXBP1-related research). Below we will discuss the respective benefits of both of these models.
Modeling STXBP1 in C. elegans
C. elegans have become a gold standard model in biomedical research as they provide an economical, rapid assay (especially when compared to mouse models, as studies in C. elegans can take a quarter of the cost and be completed in half the time as an equivalent study in mice). C. elegans have become an established model for epilepsy research in particular thanks to their high translatability to human genes (all of the major genes associated with epilepsy in humans have conserved orthologs in the worm), while having this simplistic, easy to characterize, structure [Figure 2].
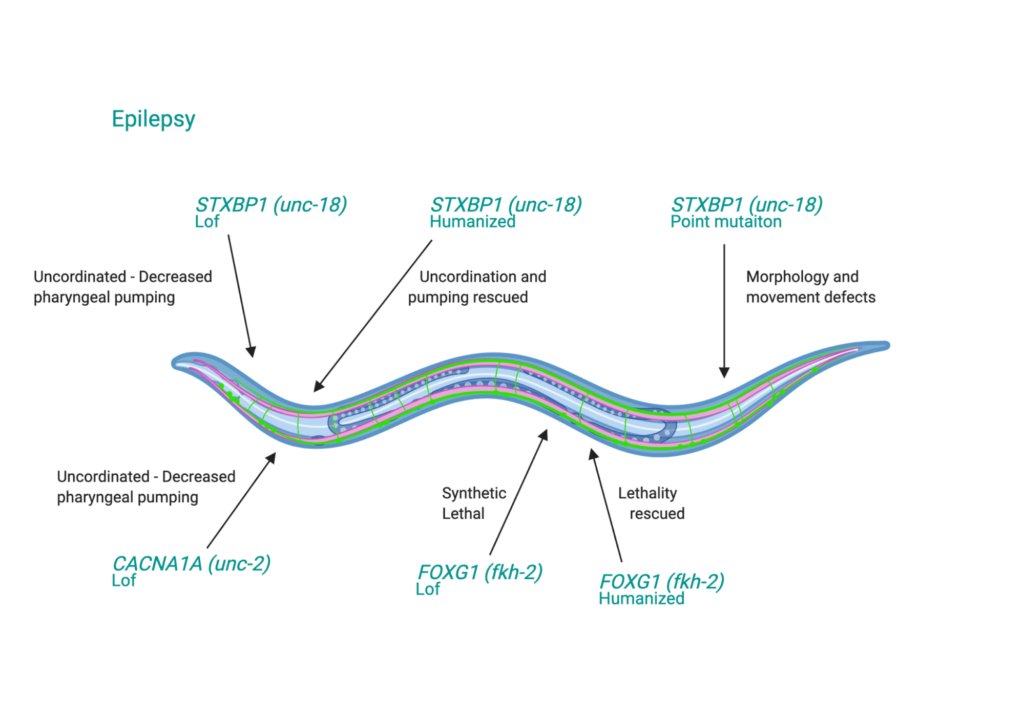
Figure 2. C. elegans as a transgenic model for studying Epilepsy Disorder (ED). Green text: ED human genes expressed with worm gene ortholog (in parentheses). Lof = Loss-of-function
Recently, the InVivo Biosystems team outlined the creation of our C. elegans epilepsy model in the publication now in print at Genetics in Medicine Open, ‘Clinical variants in Caenorhabditis elegans expressing human STXBP1 reveal a novel class of pathogenic variants and classify variants of uncertain significance’ First, the C. elegans were fully humanized, this consists of replacing the worm’s entire coding sequence with the human sequence, resulting in a model (or ‘patient avatar’) which can act as a stand-in for a patient in testing. Second, variants (known benign and pathogenic, and VUS) were installed in the humanized C. elegans [Figure 3]. Third, the animals were plated in petri dishes and their activity was video recorded to capture their behavior patterns, as categorized into 26 phenotypic features. Fourth, the behavioral data from known variants (25 pathogenic and 32 benign) was used to train machine learning classifiers to identify their correct classification. Lastly, the data from the VUS-expressing animals was given to the machine learning algorithms. The algorithms identified abnormal function in 8 missense variants – indicating that they are pathogenic.
Model Creation
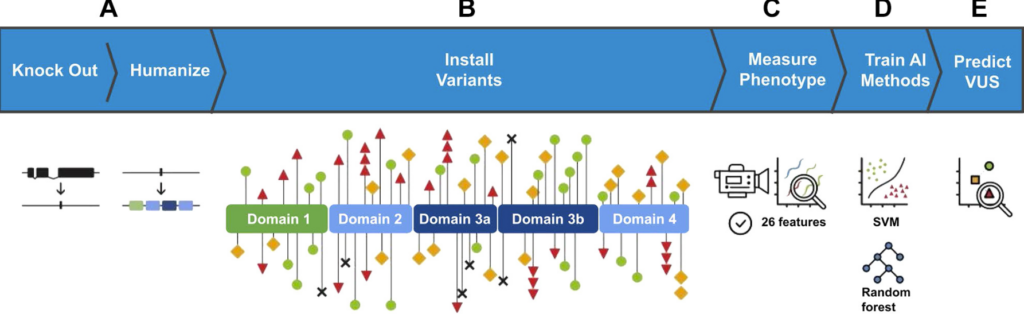
Figure 3. Schematic representation of the experiments performed. A. The native C. elegans ortholog of STXBP1, unc-18, was removed from the genome in a full deletion knockout. Subsequently, a codon optimized coding sequence encoding human STXBP1 is inserted into the same genomic location. B. Individual human variants were created in the STXBP1-expressing animals. The functional domains of STXBP1 as determined via crystallography are depicted, and the location of individual variants are marked with shapes. Red triangles represent pathogenic missense variants in our training data set, black X’s represent pathogenic truncating variants, green circles represent benign variants, yellow squares represent VUS. C. The generated strains were automatically assessed for 26 phenotypic features characterizing the animals’ movement and morphology. D. Two machine learning models, random forest and SVMs were trained on the resulting data set. E. Models were used to sort VUS into functionally normal and abnormal groups, representing functional predictors of pathogenicity. SVM, support vector machine; VUS, variant of uncertain significance.
Data exploration and feature engineering
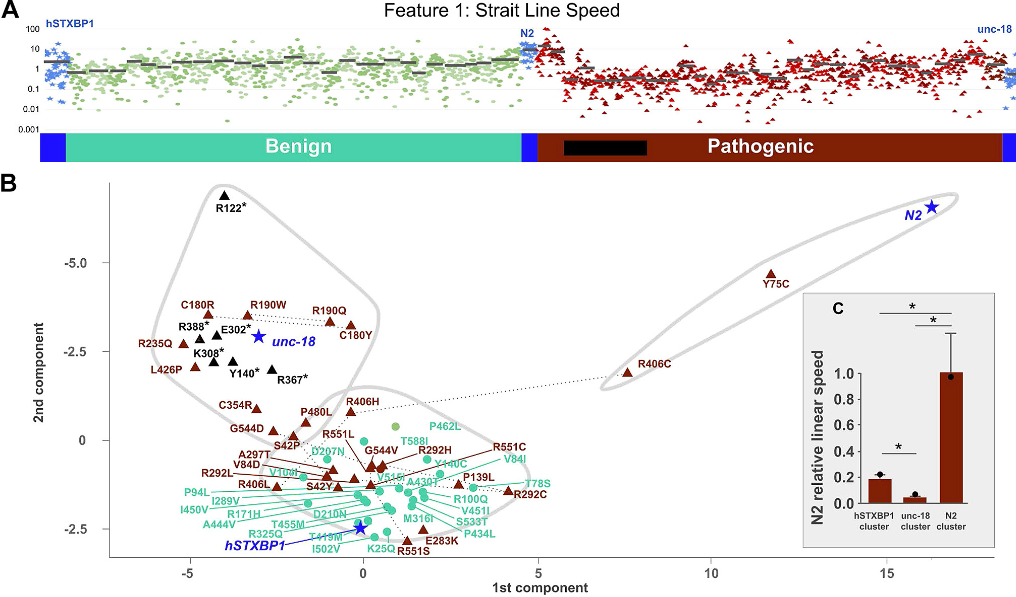
Figure 4. Example data and unsupervised cluster analysis. A. Straight Line Speed measured across 2034 worms from 60 genotypes: 25 benign (cyan) and 32 pathogenic variants (brown) plus controls (blue: unc-18 full deletion null, hSTXBP1 whole-gene humanized, and N2 wild-type) (average strain value marked by black bar). B. Pathogenic missense (brown triangles), pathogenic truncating (black triangles), benign (cyan circles), and control (blue stars) variants were clustered using k-means algorithm and plotted in 2 dimensions (principal components 1 and 2), which shows 3 distinct clusters (gray-bound regions). C. Inset bar graph shows linear speed for each pathogenic cluster, with standard error (whiskers) and speed of the control sample in each cluster (black circle). Asterisk indicates P value < .05 (t test). R:Arg; N:Asn; D:Asp; C:Cys; E:Glu; Q:Gln; G:Gly; H:His; I:Ile; L:Leu; K:Lys; M:Met; F:Phe; P:Pro; S:Ser; T:Thr; W:Trp; Y:Tyr; V:Val.
Knowing that no single feature could reliably be used to indicate benign vs. pathogenicity, the team identified 26 features. Thus, after generating the C. elegans models, the team performed k-means clustering, and notably, found that each cluster contained 1 of each control [Figure 4].
Training Machine Learning Algorithms
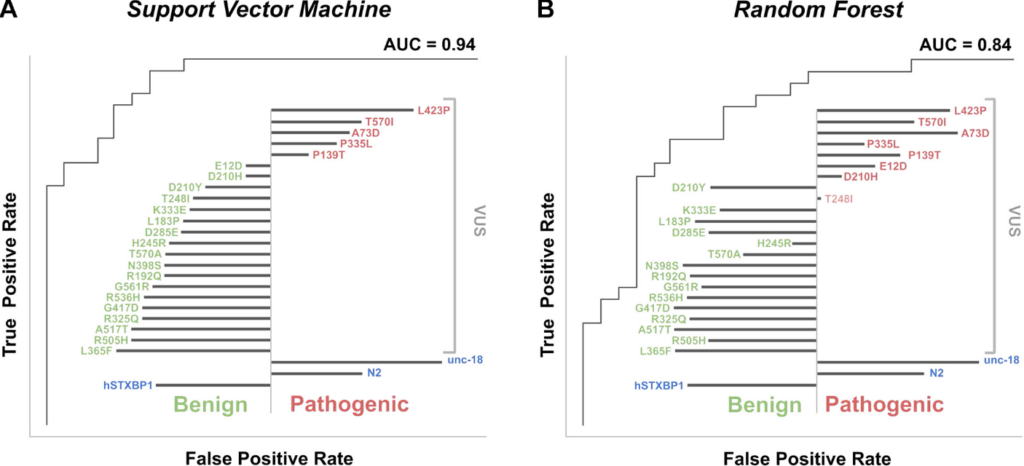
Figure 5. Model evaluation and functional predictions using supervised machine learning algorithms. VUS predicted pathogenic (red text) and benign (green text) are shown; the length of black bar (left for benign, right pathogenic) indicates strength of classification. A. A support vector machine classification, of known benign and pathogenic variants, achieved an AUC of 0.94 and classified 5 of 23 VUS as pathogenic. B. A random forest classification achieved an AUC of 0.84 and classified 8 of 23 VUS as pathogenic. R:Arg; N:Asn; D:Asp; C:Cys; E:Glu; Q:Gln; G:Gly; H:His; I:Ile; L:Leu; K:Lys; M:Met; F:Phe; P:Pro; S:Ser; T:Thr; W:Trp; Y:Tyr; V:Val. AUC, area under curve.
The team utilized two orthogonal classifier algorithms, which were in good agreement, only differing on 4 of 57 variants in the training set. There were some misclassifications (8 misidentified as benign and 3 misidentified as pathogenic), however these were located only in the Arg551 and Arg292 mutational hotspots, and the algorithms were accurate in identifying all variants at Arg406 [Figure 5]. It is possible that this is a result of an inability to detect dominant negative features, and thus, this can be refined in future work.
Overall, our approach outlined here depicts a fast, efficient platform for functional testing of epilepsy genes with the notable benefits of simultaneously being low cost while still having the benefits of a whole organism context.
This work screened 32 pathogenic variants in the STXBP1 gene, which accounts for nearly 25% of all known pathogenic variants in the STXBP1 gene. Thus, through work such as the creation of STXBP1-humanized C. elegans outlined in this paper, InVivo Biosystems is contributing to a community of researchers that are working to rapidly classify VUS.
The team has also continued this work in the other model organism we employ – the zebrafish, which we will briefly discuss below. To learn more about our work in zebrafish, read our latest blog on generating zebrafish models of epilepsy.
Modeling STXBP1 in zebrafish
When aiming to understand movement disorders caused by variants in rare diseases, such as epilepsy, it is important to have a variety of available models. To this end, the scientists at InVivo Biosystems generated a subset of the variants created in the previous body of work modeling STXBP1 in C. elegans, but this time in the zebrafish system.
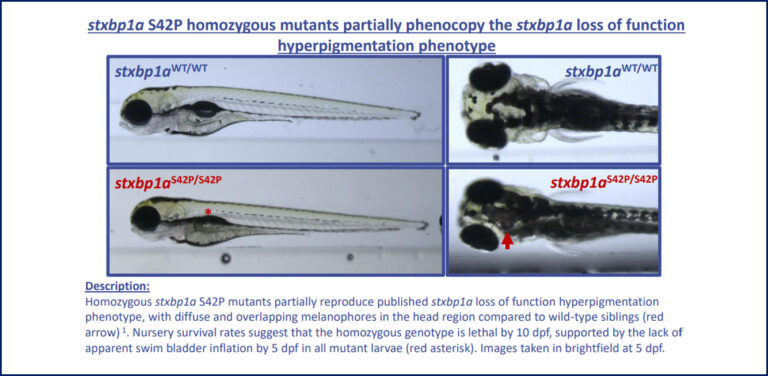
In this work, we created two point mutations in the zebrafish stxbp1a gene, a highly conserved ortholog of human STXBP1 (87% identity). Using CRISPR/Cas9 technology, the team was able to precisely generate both a predicted benign patient mutation at the conserved amino acid residue (p.P94L) as well as a predicted pathogenic patient variant (p.S42P). In this work we aimed to assess differences in phenotypes with respect to established loss-of-function mutants, which present with severe movement defects and lethality by 5 days of age in homozygous animals.
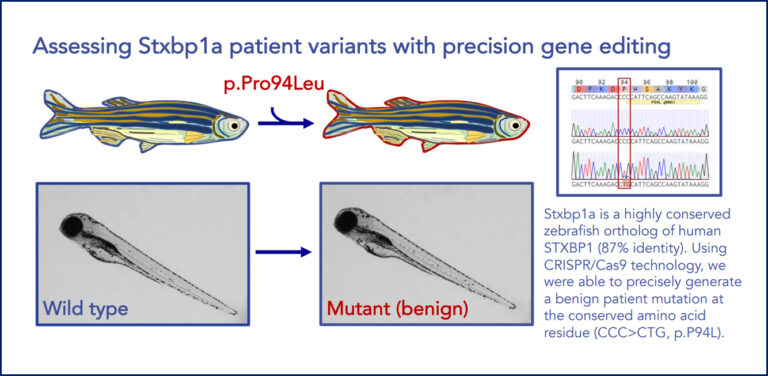
Figure 6. Illustration of a successful precise point mutation of stxbp1a in zebrafish. stxbp1a is a highly conserved zebrafish ortholog of human STXBP1 (87% identity). Using CRISPR/Cas9 technology, we were able to precisely generate a benign patient mutation (above; p.P94L) and a pathogenic patient mutation (below; p.S42P) at the conserved amino acid residue (p.P94L).
To learn more about our epilepsy work using zebrafish, and how we can advance your project, talk to one of our experts!
Conclusion
While the work here describes understanding variants in STXBP1 specifically, it is meant to demonstrate how organisms such as C. elegans and zebrafish can be effectively used to model many such genetic epilepsies. With the huge number of VUS being identified through genomic and predictive measures, it is important to have easy to use in vivo models that help us understand the criticality of any individual variant or combination of variants and their impact on disease.
References
- Green, Eric D., et al. “Strategic vision for improving human health at The Forefront of Genomics.” Nature 586.7831 (2020): 683-692. https://doi.org/10.1038/s41586-020-2817-4
- Hopkins, Christopher E., et al. “Clinical variants in Caenorhabditis elegans expressing human STXBP1 reveal a novel class of pathogenic variants and classify variants of uncertain significance.” Genetics in Medicine Open 1.1 (2023): 100823. https://doi.org/10.1016/j.gimo.2023.100823
- Gunning AC, Fryer V, Fasham J, et al. Assessing performance of pathogenicity predictors using clinically relevant variant datasets. J Med Genet. 2021;58(8):547-555. http://doi.org/10.1136/jmedgenet-2020-107003
- CHOP (2021). STXBP1-Related Disorders. Children’s Hospital of Philadelphia. https://www.chop.edu/conditions-diseases/stxbp1-related-disorders
- STXBP1 Foundation (2021). About STXBP1. STXBP1 Foundation. https://www.stxbp1disorders.org/what-is-stxbp1

