

Dravet syndrome is a rare but severe form of epilepsy that begins in infancy, challenging families and neurodegenerative disease researchers alike to find effective treatments amidst a landscape of uncertainty. In the pursuit of hope, the unassuming zebrafish offers unique advantages for understanding and treating this debilitating condition. This post highlights the growing role of zebrafish in epilepsy research, highlighting their potential to accelerate the discovery of life-changing medications.
Key Points
- Dravet syndrome is a rare and treatment resistant form of epilepsy.
- Zebrafish with mutations in the gene scn1lab effectively model Dravet syndrome, with key advantages over existing rodent epilepsy models.
- The RapidGen™ scn1ab zebrafish can be quickly generated to enable rapid variant assessment or early drug screening.
What is Dravet Syndrome?
Dravet syndrome is a treatment-resistant epilepsy disorder of genetic origin. Seizures begin in the first year of life, often brought on by fever (febrile seizure), though subsequent seizures do not require a fever. Once seizures develop, children affected by Dravet syndrome are also likely to experience developmental delays, movement and balance issues, sleep disturbances, and autism-like features.
The vast majority of people with Dravet (~90%) have a mutation in the gene SCN1A, which encodes a voltage-gated sodium channel subunit. Voltage-gated sodium channels are found on cell membranes and mediate sodium flow into cells, which is critical for communication between neurons in the brain. Disruption of voltage-gated sodium channels like SCN1A can cause seizures by increasing neuronal excitability.
Most people with Dravet syndrome have a de novo SCN1A mutation that was not inherited from their parents, who would therefore not have Dravet syndrome themselves.
Dravet syndrome seizures are difficult to control with existing therapeutics. Current treatments may include a combination of anticonvulsant medications, ketogenic (low carbohydrate, high fat) diet, and other interventions including cannabidiol (CBD), vagus nerve stimulation, and cognitive therapies. Due to the lack of a broadly effective treatment paradigm, discovery of additional drugs that may help control Dravet syndrome seizures is critical.
Why are Zebrafish an ideal system to model Dravet?
- The zebrafish scn1ab gene is 77% similar to the human gene for modeling Dravet syndrome.
- Zebrafish seizures are easily detected in well plate formats, providing a reliable and scalable way to test a large number of potential therapeutics.
- The RapidGen™ scn1ab zebrafish are generated in 2-7 days, followed by screening for therapeutic rescue within another 3-7 days.
Though mammalian models have been used to study Dravet syndrome, several features of these models have limited the discovery of new therapeutics. For example, the challenge of breeding mouse models and strong influence of background genetics on epilepsy phenotypes can hamper high-throughput, replicable studies. Induced pluripotent stem cells (IPSCs), which have been used to model individual patient mutations in SCN1A, cannot recapitulate the complexity of brain function that generates seizures in Dravet syndrome patients.
Zebrafish have many advantages for studying Dravet syndrome and identifying new therapeutics. First, seizures are easily detected in zebrafish by measurement of distinct locomotor activity, including burst swimming, clonus-like like convulsions, and loss of posture.
These behaviors are easily assayed in hundreds of zebrafish from a single clutch, in well-plate formats that facilitate high-throughput screening of potential therapeutics in a whole organism system. Large clutch sizes also make it easier to control for background genetic effects by assaying genetically similar siblings. Larval zebrafish are also small and transparent, which makes it easy to study how therapeutics impact brain structure and function in large numbers of intact animals.
Zebrafish have already been a productive model for identifying Dravet syndrome treatments; serotonin-modulating compounds including clemizole, trazodone, and fenfluramine (which is now an FDA and EU approved intervention for Dravet syndrome) are among those that suppress seizures in zebrafish Dravet syndrome models.
See our epilepsy model page for more information about our data insights
Epilepsy Modeling Services
How does the IVB team model Dravet?
Zebrafish have two genes that encode a voltage-gated sodium channel subunit analogous to human SCN1A: scn1laa and scn1lab. Conveniently, the zebrafish genome duplication means that homozygous loss-of-function mutation in scn1lab has been found to effectively model features of SCN1A haploinsufficiency in Dravet syndrome. Zebrafish scn1lab is also 77% identical to human SCN1A, making it possible to model hundreds of individual patient SCN1A variants in zebrafish.
At InVivo Biosystems, we use CRISPR/Cas technology to generate scn1lab mutants and model Dravet syndrome. In our RapidGen™ approach, optimized CRISPR reagents enable us to generate hundreds of scn1lab mutants in a matter of weeks for application in diverse downstream experiments. For example, we can take a personalized medicine approach using mRNA microinjection to supplement scn1lab mutants with specific patient variants, or we can generate stable mutant lines that can be applied in larger format screening. Libraries of natural or pharmaceutical compounds can then be applied to any of these Dravet syndrome models and screened for their ability to restore Dravet-related phenotypes.
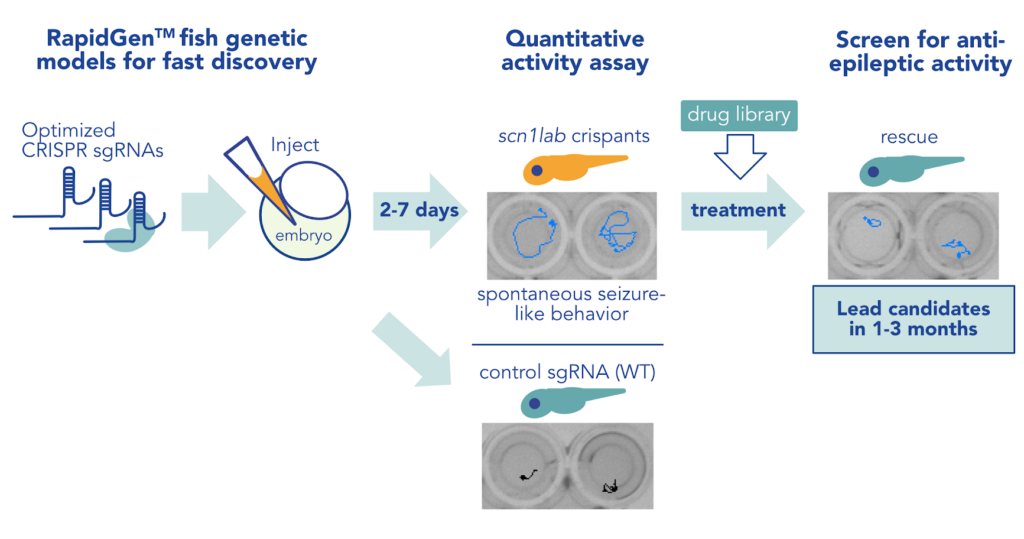
How do we assay Dravet syndrome phenotypically?
We have the capability to assay many Dravet-related phenotypes in scn1lab mutant zebrafish. For example, seizure-like movement, circadian rhythm disruption, altered brain electrical activity, and metabolic changes have all been detected in zebrafish Dravet syndrome models. For high-throughput screening of potential therapeutics, our Seizure-like Movement Assay uses high-resolution video tracking of zebrafish movement to identify seizure bouts in scn1lab mutant larvae. We can also use our instrumentation to customize seizure-inducing stimuli or behavioral endpoints to screen for compounds more likely to act on specific mechanisms. By quantifying seizure bouts in dozens of zebrafish simultaneously, we can generate datasets that include up to 50 animals in each experimental group and therefore provide superior confidence in the positive hits we reveal.
Critically, the ability to easily assay multiple Dravet-related phenotypes in zebrafish also enables us to avoid false-positive hits, such as compounds with sedative effects that don’t affect seizures, that can be abundant in simpler screens in other models.
Conclusion
Rare diseases such as Dravet syndrome exemplify the need to both develop relevant models as well at accelerate the pace of therapeutic discovery. At InVivo Biosystems we are drawing from both our cutting-edge genome editing techniques as well as our deep body of work in epilepsy to find novel therapies for drug-resistant seizures in Dravet syndrome. Recent advances have made it abundantly clear that innovative small animal models such as zebrafish will play an expanding role in confronting rare diseases.


