
Just the other day, I was talking with a collaborator who told me that they didn’t really believe that knock-ins really worked in zebrafish. Although there have been literature reports for years, the number of labs that struggle and fail with this technique far outweigh the number that find success and publish. So I can forgive my colleague for being a little doubtful when I mentioned that we can successfully make knock-in transgenics. “Do you actually have preliminary data that you can make CRISPR knock-ins?” She asked me point blank. “I know many groups trying, but I don’t know of success.”
I started asking around about how easy it is to make knockins in Zebrafish. The handful of people contacted said they knew it “to be hard but that there are published reports coming out saying it is possible.” So why do so many zebrafish groups struggle to achieve knock-in transgenesis? What is so hard about knock-ins? To start to understand, let’s talk a little more about CRISPR, the technique that makes knock-ins possible.
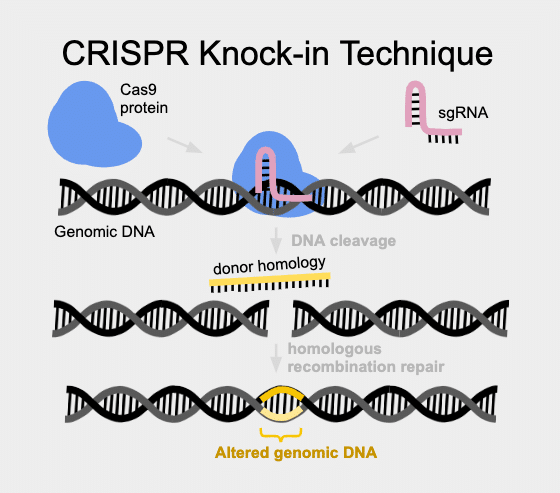
CRISPR-based gene editing can result in either knock-ins or knock-outs, taking advantage of a variety of inherent DNA repair mechanisms. Non homologous end joining (NHEJ) is the cell mechanism to rescue breaks in DNA in a sloppily imprecise way. In contrast, Homology Directed Repair is much more faithful and can be harnessed to put specific content into a cut site.
Knock-ins use Homology Directed Repair (HDR) and can be used to create very specific changes in the DNA sequence. These changes can be as small as only one base pair of DNA (also known as a point mutation), or as large as an entire protein, (as in GFP, LoxP sites, and more). HDR uses a donor homology DNA template to instruct precision gene repair from the double stranded break induced by Cas9. One group reporting a high success rate with large knock-ins is the David Grunwald Lab at the University of Utah in Salt Lake City. According to this group, the key is to design the donor homology DNA to contain at least 1000 base pairs on each side of the insertion cargo (PMID 27003937). In this method, a plasmid frequently needs to be created containing homology arms and cargo content.

For smaller precision edits, the use of oligonucleotides, or ssODNs is called for (PMID: 23000899). An ssODN (which is essentially a short single-strand piece of DNA) can be made very easily and quickly, using any number of synthetic oligonucleotide suppliers. These short pieces of single-strand DNA are often referred to as ssODNs. When an ssODN is synthesized for use as a template to instruct precision DNA repair, it can be put into an injection mix containing Cas9 and sgRNA. This targets a break at the sgRNA encoded site and the ssODN “instructs” the cell’s natural DNA repair machinery to fix the break and put in the ssODN (“donor homology”) content. The exact optimal length of ssODN to use for optimal efficiency of edits is still debated, but seems to be between 25-50 base pairs.
So what biological mechanisms do these two types of homology directed repairs use? When homology arms are long (+500bp) and double stranded, it is classical homologous recombination repair with Holliday junction formation. When donor homology is single stranded a Holliday junction cannot be formed. Holliday junction formation is the singular trademark of homologous repair, so something else must be taking place. What seems to be happening is single strand annealing (SSA), akin to what goes on in Base Excision Repair. Here, a strand of DNA invades and is used as a template for repair. This is not too far removed from holiday junction, but it is just one sided – there is no invading complementary strand.
CRISPR knock-outs are a little more messy and unpredictable. Donor homology is absent, so repair of a DNA break is left to the faulty DNA repair pathway of non-homologous end joining (NHEJ). This method has a high propensity to make mistakes that lead to new genetic information, often as an insertion or deletion (indel) causing a frame-shift which leads to incomplete protein being made. These protein truncation variants (PTVs) are almost always highly disruptive and lead to loss of function in the shortened stub of protein that gets made.
(image: NHEJ and HDR) (Ramon Guitart et al., 2016: “Research Techniques Made Simple:The Application of CRISPR-Cas9 andGenome Editing in Investigative Dermatology” http://dx.doi.org/10.1016/j.jid.2016.06.007)

Competing Repair Mechanisms of NHEJ and HDR
So why do Knock -Ins continue to be difficult in Zebrafish, given that we know so much about the mechanisms at work? One reason is that even when we scientists are putting in all the templates needed for HDR, the NHEJ mechanism is still highly active in the cell. The NHEJ mechanism competes against our desire to insert precision cargo content and instead makes semi-random insertion/deletions (“indels”) after the Cas9 cuts the DNA. In many donor-homology templated gene-editing experiments, the creation of indels occurs at a very high rate and a set of embryo injections will yield many more indels than targeted edits. Often those indels disrupt coding, so if the gene is essential, the embryo will die fairly quickly due to high efficiency of Cas9 cutting leading to a majority of the embryo cells (both “soma” and “germline”) getting indels that cause abnormal production of the gene’s protein.
Today’s and Tomorrow’s Knock In Efficiency
As a community, we appreciate that getting CRISPR based knock-ins to occur efficiently in zebrafish is quite challenging. We conducted a survey of zebrafish researchers, and all respondents agreed that getting knock-in transgenics is extremely difficult. Our team has found success can be higher with well designed reagents and processes. We are approaching an 80% success rate in the knock In projects we have tried. Yet we need improvements in the process so that everyone can get access to higher efficiency knockin transgenic production.
Are you looking to advance your research and explore the possibilities of genome engineering in zebrafish? Our zebrafish CRISPR Knock-In Service offers a cutting-edge solution for precise genetic modifications, allowing you to achieve targeted gene insertions with high efficiency and accuracy.
Learn more about InVivo Biosystems’ zebrafish knock-in services.


