
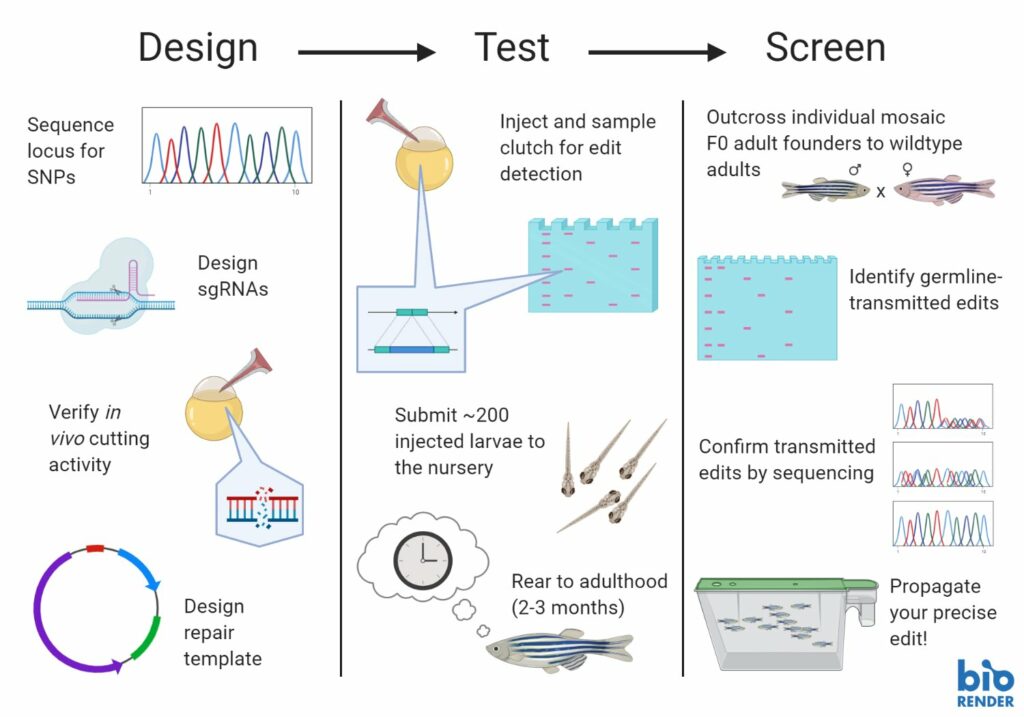
Knock-ins are hard, for a long time people even said they were impossible, but we are nearing an 80% success rate for CRISPR knock-ins in zebrafish so we want to share with you what we believe to be the 11 steps necessary to have a successful knock-in project.
1. Know your gene.
Good knowledge of your gene, alternate isoforms expressed and related orthologs is ideal for designing an efficient experimental strategy. Your wild-type zebrafish strain may differ from the reference sequence, so you may want to sequence your gene in your strain to ensure that you know its precise DNA sequence.
2. Design sgRNAs.
Multiple sgRNAs with different recognition and PAM sites should be designed for each knock-in experiment to ensure you use an sgRNA that guides efficient cutting. When choosing sgRNAs, the cut site should be as close to the knock-in site as possible to increase edit efficiency. Be sure to check for potential unwanted off-target effects predicted in your sgRNA design algorithm.
3. Validate sgRNAs.
All sgRNAs should be validated in vivo to ensure that they are not toxic and that they efficiently guide Cas9 cutting of the DNA. Injections must be performed on healthy embryos within 1 hour of fertilization. Cutting can be observed by a disruption in a sequencing read. Quality PCR amplification and DNA sequencing is required to be able to detect this disruption.
4. Design donor DNA.
There are two types of DNA donor templates: plasmid templates, which are double-stranded DNA (dsDNA), and single-stranded oligonucleotide DNA (ssODN). The size of your knock-in insert determines the type of template and the size of the homology arms used. The homology arms flank the knock-in insert sequence. To prevent recutting by Cas9, silent mutations should be introduced in the donor template so that the sgRNA no longer recognizes the target site or that the PAM site is eliminated.
")
5. Embryo injections.
Microinjections must be performed on healthy embryos within 1 hour of fertilization. Because CRISPR knock-ins are relatively low efficiency, as many as 600 embryos may need to be injected with a 95% survival rate to achieve your edit of interest.
6. Rear your F0 zebrafish.
Take good care of your fish! You won’t know if your edit was successful until they reach adulthood and can produce embryos. Ensure that tanks are well maintained and fish are consistently cared for.
7. Identify F0 founders.
Developing a robust assay to detect your edit is critical when you begin screening the embryos produced by the F0 founders. Three possible methods include mutation-specific PCR, high-resolution melt analysis (HRMA), and restriction enzyme digest screening.The F0 germline is mosaic and can transmit multiple edits, and you can miss your edit if you don’t have a good assay to detect it. With knock-in efficiencies ranging from 0.1-9% in the literature, expect that you may need to screen embryos from many fish.
8. Sequence the transmitted edit.
Perform Sanger Sequencing or NGS to confirm that your edit occurred as expected with no extraneous sequence added. Erroneous integration at the site of the double-strand break may screen as positive in your F0 assay. A recent paper found that the rate of perfect repair dropped from 4-8% down to 1-4% after removing erroneous integration events (Boel et al, 2018).
9. Rear your F1 heterozygotes.
The rate of transmission to your F1 generation will vary depending on when the F0 germline edit was made in development. Ranges from 3% to 50% have been observed. Be sure to grow enough F1 animals that you will be able to find your F1 heterozygotes.
10. Finclip to identify F1 heterozygotes.
Caudal fin genotyping can be useful to identify candidate heterozygotes. At this stage, the edit is not mosaic and every cell of the fish should have the same genotype.
11. Incross F1 heterozygotes to find F2 homozygous fish.
In order to get a stable, homozygous line, the heterozygotes must be crossed together. The resulting F2s may again be assayed via finclip to find the homozygotes. Sequencing of the homozygous fish is a good confirmation that you have the correct edit. You may also consider a Western blot or immunostaining with a validated antibody to characterize the protein product of the genome edit.
Now your transgenic fish are ready, and you can do your real experiment!

