
A Question One Might Ask:
How do you make animal models more effective at drug discovery?
One process, now made simple using CRISPR technologies, is to replace genes in the animal with human genes. Surprisingly, we have found gene substitutions can be made where the human coding sequence can be used to restore loss of function in important disease associated genes [1]. This is a big step forward from the standard way animal models are used.
The traditional approach of looking at precision medicine in animal models has two major limitations:
2. Drug Binding Sites are Faulty. Amino acids between the human and animal model are rarely identical and the drug binding site topology is often not conserved between the species.
However, we can effectively remove these concerns from the drug discovery equation by using animals that are humanized for their drug targets. A major concern with humanization is the ability to have enough sequence identity so that a human gene can retain function when inserted as a gene substitution. Fortunately, the detection of gene-to-gene homology (quantified number of genes with at least 25% sequence identity) is quite a large fraction of disease-associated genes. For example, when we compare the genomes of C. elegans animal model (“the worm”) to humans, and compare the zebrafish genome to human, we see that many of the important gene associated with disease are shared between the models and humans (worm = 84% and zebrafish = 98%) (Figure 1).
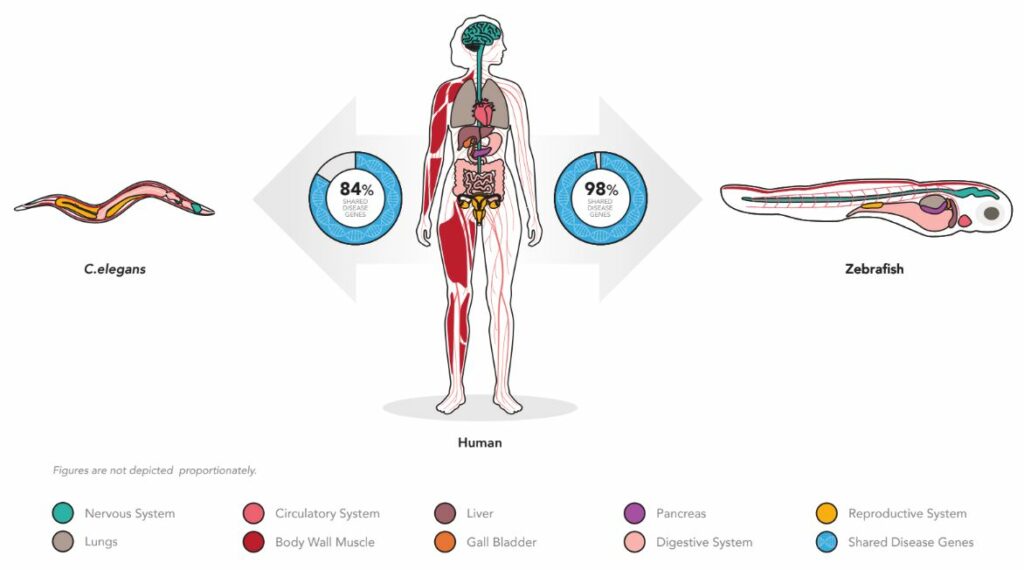
Figure 1. Both the C. elegans worm and the zebrafish have significant number of shared genes and tissue othology allowing for high face validity in data translation to clinic
This phenomenon of sharing important disease-associated genes (along with the two major limitations listed above) lead us to develop the Whole-gene Humanized Animal Model (WHAM) platform. Using components of multiple NIH grants totaling $1.9 million, InVivo Biosystems started to develop the WHAM procedure in mid 2010’s. First, human transgene expression was done from safe harbor sites using transposon mediated gene insertion. Next, direct replacement techniques were used to swap in human coding sequence for the animal’s coding sequence. The culmination of this work resulted in a recently awarded patent (US 11477970 B2).
Background -Why are alternative drug screening platforms needed?
In silico methods are robust for target-specific discovery of therapeutics [2]. However this method reaches a disconnect when validation of drug candidates is performed in standard (unmodified) animal models. This disconnect occurs because drug binding sites are often faulty – the in silico drug design occurred on a protein that is an exact virtual copy of the human coding sequence, however the screening of candidates for activity is performed on the animal’s version of the target protein, which is rarely an exact copy of the human coding sequence. Thus when looking at a patent variant inserted into the animal’s version of the disease gene, there are a high number of false positives (drugs acting on animal gene but not active on human gene) and high number of false negatives (drugs that would have acted on human gene but are missed because they don’t act on the animal gene). The WHAM-humanization platform offers a solution to this dilemma because it uses the same human coding sequence as was used in the in silico work. As a result, the WHAM-humanization method is a promising way forwards in new drug development.
Example of the benefit of WHAM in CF gene
In silico methods’ success in the rare disease research space. One of the first discovered genes in Rare Disease was the Cystic Fibrosis, the CFTR gene. Drug development in this genetic deficiency has recently seen the discovery of highly successful therapies [3,4]. With polytherapeutics identified as the next evolution in drug development [5], a highly successful drug combination for treating Cystic Fibrosis was achieved with the development of Trikafta [6]. A three component mixture has two components functioning independently to stabilize the CFTR chloride channel so that it can be transported to the plasma membrane (Elexacaftor and Tezacaftor). The third component acts to modulate ion permeability so that once the channel is at the plasma membrane, it can experience an increased residence time in open channel state (Ivacaftor). Molecular dynamics studies have been used to identify two binding sites for one of the stabilizers (Tezacaftor), which occurs in a thermodynamically unstable region of CFTR transmembrane domain [7]. Subsequently, it was found via cryo-EM structures that indeed, Tezacaftor binds to one of these sites in a binding pocket that allows the compound to insert between the transmembrane helices of CFTR and the resulting stabilization allows the channel to continue proper processing and be trafficked out to the plasma membrane [8].
Where WHAM-humanization can help. The residues of Tezacaftors binding site have been mutated to eliminate charge or contain bulky side chains that interfere with drug binding. The result of these binding pocket mutations is Tezacaftor can no longer promote proper trafficking out to the plasma membrane [7,8]. For instance, the arginine at R74 when mutated to an alanine leads to blockade of Tevacafor’s protein stabilization activity and the channel no longer makes it out to the membrane. When these residues are mapped to the ortholog sequence of various model systems, we observed that both zebrafish and C. elegans, lack complete conservation in these essential residues (Table 1). In an extended analysis the 22 amino acids involved in Tezacaftor binding were examined across species and amino acid conservation per animal was 77% (mouse), 36% (fish), and 40% (worm). These results suggest there is a necessity of using a Whole-gene Humanized Animal Model (WHAM) to replace the animal’s ortholog with human coding sequence to enable a screening platform capable of detecting human relevant therapeutics.
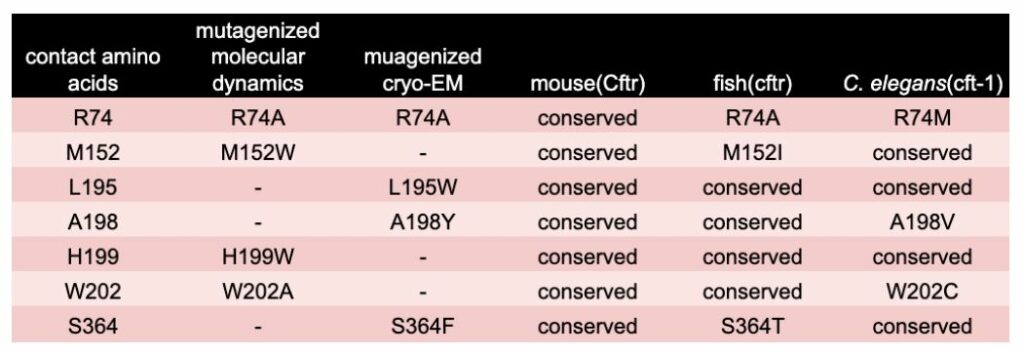
Table 1. Conservation of Amino Acids at Tezacaftor’s Binding Site
WHAM humanization results in human genes that can rescue loss of function defects. At InVivo Biosystems, we have performed the WHAM procedure (Figure 2) at multiple gene loci and observed over 80% of the gene substitutions lead to significant rescue of gene function [1]. In the WHAM approach, it is necessary to alter the coding sequence of the inserted human gene in order to optimize expression in the animal model. For instance, the human genome coding sequence is 15% richer in GC content when compared to C. elegans [9]. This can create a codon bias where certain codons that are abundantly used in the human genome are observed as rare in the C. elegans genome. To avoid the usage of rare codons in C. elegans, codon adapters are available to enable selection of more frequently used codons [10,11]. A second attribute to enable adequate expression of the human coding sequence is to introduce artificial introns, which helps avoid triggering of the nonsense-mediated-decay (NMD) mechanism that leads to degradation of intronless transcripts [12,13]. A third attribute of design is to detect and eliminate cryptic splice junctions via sequence scanning software (e.g. the NetGene2 program) [14,15]. The final attribute that measures the success of a humanized sequence is the observation of rescue-of-function relative to a knockout or loss-of-function allele in the parent line. A whole-gene humanized platform can be considered as valid for use, if statistically significant restoration of function can be achieved with the wildtype human coding sequence.
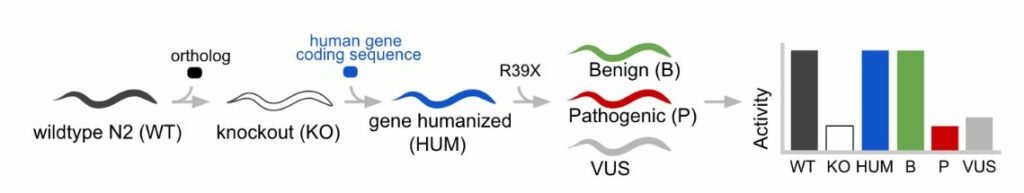
Figure 2: Outline of the Whole-gene Humanization in Animal Model (WHAM) approach in C. elegans. A native (WT) animal is CRISPR-edited to make a gene KO animal. A whole gene substitution is made with a human coding sequence in the native locus. Next, additional CRISPR edits are made to insert clinical variants (eg “R39X”) into the humanized locus which recreates a set of Patient Avatars that represent the genetic variation of a human. Phenotypic activity of the transgenic lines is measured to enable detection of variant deficiencies.
InVivo Biosystems’ Approach to Creating a Drug Screening Platform
In general, the approach we take utilizes a combination of in silico simulations of molecule binding to find hits and then test the hits for activity in a WHAM-humanized animal model. The approach is iterative and uses data from hit activity as feedback into AI-driven binding simulations. This recycling back to the in silico then derives new scaffolds with ever tighter binding affinities to the gene target. Unlike many gene-associated diseases, a large portion of the VCP variants are pathogenic via gain-of-function (GOF) mechanisms [16,17], and therefore, inhibitors of this enzyme’s function are likely to be therapeutic. However, it also has been noted that a small proportion of VCP variants are loss-of-function (LOF) [18], and therefore, activators of protein function or stability may be therapeutic. As a result, our approach will focus on discovery of ligands that act on both the enzymatic catalytic site and at thermodynamically vulnerable hotspots within the protein. Our approach will be carried out in three aims:
In Silico AI-driven Drug Discovery, Design and Development. Prior work has identified a variety molecules that can acts as inhibitors of VCP where their scaffolds range from Dibenzylquinazolines, Alkylsulfanyl-1,2,3-triazoles, Tetrahydrocarbazoles, Pyrazolo [3,4-d] pyrimidines to Curvularins and their binding sites range from direct inhibitors to allosteric modulators [19]. These scaffolds and other AAA ATPase interacting molecules will be used as starting places for discovery of unique compound architectures. We will derive a composite molecular structure, comprised from multiple x-ray sources, Cryo-EM and NMR. Next we will use structure-based drug discovery to develop and refine exact pharmacophores for 3D-QSAR with machine learning to screen against ~2 billion molecules that will identify high-affinity ligands. This aim combines several steps: 1) AI-based homology modeling to build absent atoms with given protonation (pH); 2) molecular mapping to identify binding hot spots on protein’s surface; 3) massive molecular docking with around 2 billion compounds; 4) advanced molecular dynamics simulations (as needed) for predicted binding affinity (dG); 5) selected compounds recalculations with flexible groups in active site of protein. Expectations for variants for VCP to be screened for groupings of compounds that treat either GOF or LOF deficiencies.
In vivo Animal Modeling for Compound Validation and Efficacy. Gene-humanized C. elegans and zebrafish are created and designed to contain a clinical variant. The resulting animals are then characterized for the presence of phenotypic defects. In C. elegans, clinical variants are created as genomic integrants in a WHAM-humanized locus expressing full length human VCP (uniprot: P55072; 806 aa), which is a gene replacement for the C. elegans ortholog (cdc-48.1; 809 aa). The likelihood for success is high because the sequence identity between C. elegans and humans is 77%, which is nearly 2 x higher than the 40% identity threshold for humanization as determined in our prior work [1]. The phenotypic consequence of loss of function in the C. elegans cdc-48.1 gene is a mild reduction in fecundity, however there is a duplicate gene (cdc-48.2) that also needs to be disrupted with a coding sequence deletion to result in non-viable animals. As a result, we will use deletion of both cdc-48.1 and cdc-48.2 loci to generate a background that can be hypersensitive to changes in AAA ATPase activity. However full genetic knockouts of these genes are not yet available, thus CRISPR-mediated deletions of the entire coding sequence of these genes will also be made. The strains to be made are listed in Table 2. For zebrafish a genetic knock down approach is initially used. CRISPR reagents injected into embryos can have significant entrance into all tissues (germline and soma). The result is often a pronounced phenotype (a “crispant“) in most of the embryos when an important disease gene is being examined. We will use this knockdown approach in zebrafish to uncover the morphological and behavioral defects that occur with loss of function in the zebrafish VCP ortholog (vcp). Rescue of the knock down is then performed with mRNA coding for the human wildtype VCP, or a VCP with clinical variant (p.VAR). These animal models systems are considered valid once three conditions are met: 1) Gene knockout or knockdown has significant LOF defect, 2) human wildtype coding sequence can rescue the LOF, and 3) a variant installed in humanized VCP sequence provides a detectable functional defect relative to wild type VCP sequence.
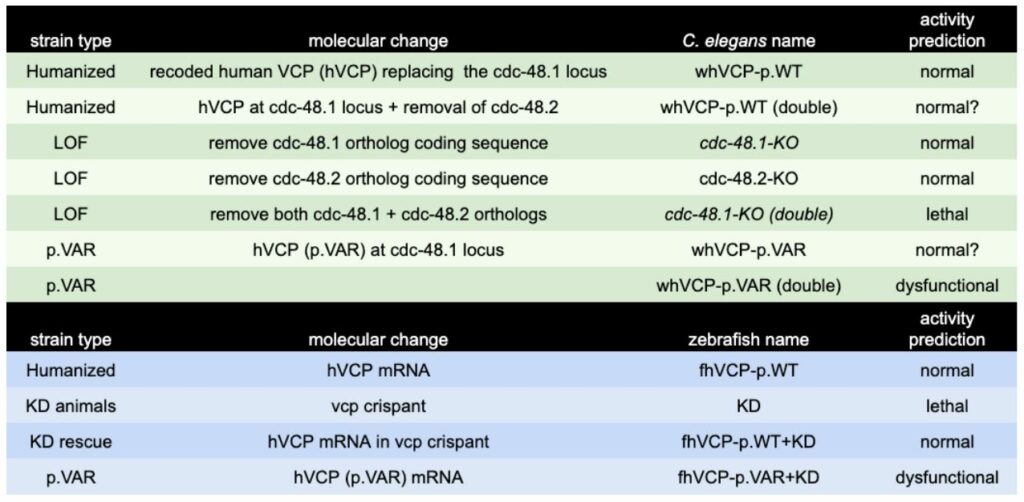
Table 2. Genetic Strains Made for VCP Project.
The most common pathogenic variant in VCP is p.R155H [20]. However there are another 21 missense variants reported in ClinVar that have pathogenic association with Frontotemporal Dementia [21], many of these occur in the N terminal domain that regulates access to ATP-binding D1 domain (Figure 3A). In fact, two hot spots of pathogenic activity occur with amino acid substitutions between N91 to D98 or R155 to R159 in the N terminal domain [21], which, along with other pathogenic variants in the N-terminus domain, can be cross reference for their conserved positions in C. elegans and zebrafish (Figure 3B), indicating that for C. elegans, imperfect conservation occurs in hot spot 2 and other regions. Most of the pathogenic variations in VCP are considered to be GOF and can directly influence the distal D2 domain’s catalytic ATPase activity in biochemical studies, with highest activity observed in R155C and A232E variants [22]. Conservation of physical binding partners has been examined (Figure 3C) and C. elegans retains 78% of these interactions, while zebrafish retains 100% of the binding partners. These results of binding partner homolog assessment suggest a high degree of functional conservation can be expected when modeling VCP variants in C. elegans and zebrafish.
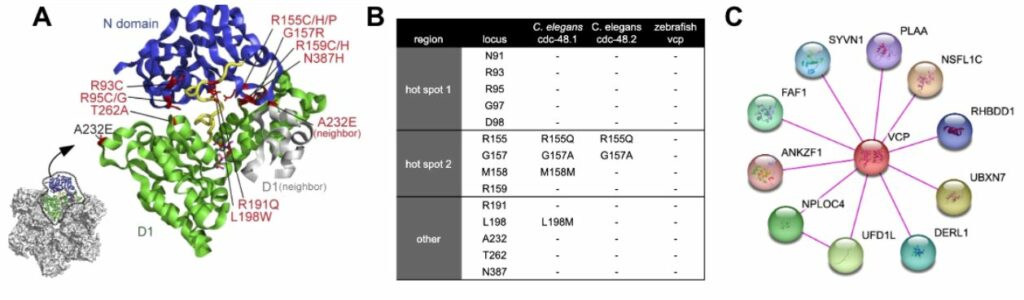
- Figure 2. A) VCP monomer truncated to N and D1 domains showing pathogenic variant hotspots (adapted from 22270372). B0 conservation of pathogenic variants in N domain from humans to C. elegans and zebrafish orthologs. C) physical interactions of VCP with other proteins (STRINGS database 33237311).
A variety of functional defects have been reported for various VCP alleles of C. elegans, zebrafish and mice. In C. elegans LOF in either a cdc-48.1 (tm544) or cdc-48.2 (tm695) loci has a mild effect of reduce brood size and slower growth, however when both alleles are combined into one animal, the result is embryonic lethality [24,25] and degradation of neuronal proteins [26]. Similarly in zebrafish, which has only one ortholog, the vcp gene, the effect of a morpholino knockdown of this gene’s expression resulted in lethality for a majority of the embryos and the defects observed were in neuronal outgrowth and increased neurodegeneration [27]. Finally in mice, a knock-in mutation is available for the human variant p.R155H where in the R155H/+ animal, the defects observed are muscle myopathy, brain aggregate formation, and motor neuron degeneration. As a result, disruption of normal activity of corresponding genes of all three animals result in similar neuronal defects.
Neuronal deficits often result in locomotion defects. We will explore a range of locomotion assays starting with simple liquid thrashing assay using a wMicrotracker apparatus to more complex video tracking of a variety of locomotion parameters using an MBF apparatus, both of which are routinely used in house. All of these assays can be performed in multiwell formats which allows for rapid assessments in these plate-reader-accessible formats.
Iterative Cycling (“Turn the Crank”). Applying the in silico discovery (computational) with in vivo validation (wet lab) allows the hits found with molecular dynamics to be tested for activity in the humanized animal models. As the catalog of compounds verified for phenotype modulation in the humanized models grows, the scaffolds of the verified hits will be examined to identify if particular theoretical pharmacophore relationships can be supported by our in vivo data. These verified pharmacophore relationships are then brought back into the molecular modeling to identify new clusters of molecules that can be tested in silico for binding and then testing in vivo. Each round of in silico with animal model validation is expected to yield compounds with increasing activity (potency) while maintaining high specificity (minimized off-target effects). We will use AI-based active learning to develop these scaffolds which will enable identification of a broad range of active and novel structures, which will enable robust intellectual property claims to be made on a variety of compositions of matter.
Discovery System Summary
We combine an in silico (computational) and in vivo (wet lab) approach in an iterative fashion to derive high affinity ligands for altering VCP stability and activity. The method requires derivation of an accurate ensemble structure of VCP and the creation of WHAM-humanized animal models that can allow bulk screening of large numbers of compounds that are refined down into a ranked list of leads verified via biological phenotype screens. The expected result upon completion of the project is a set of leads starting in the low nM affinities and high specificity through biological elimination of off target toxicities.
Learn more about InVivo Biosystems’ ability to generate models for compound efficiency and drug development here.
References:
- Hopkins CE, Brock T, Caulfield TR, Bainbridge M. Phenotypic screening models for rapid diagnosis of genetic variants and discovery of personalized therapeutics. Mol Aspects Med. 2022; 101153.
- De Vivo M, Masetti M, Bottegoni G, Cavalli A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J Med Chem. 2016;59: 4035–4061.
- Lopes-Pacheco M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front Pharmacol. 2019;10: 1662.
- Capurro V, Tomati V, Sondo E, Renda M, Borrelli A, Pastorino C, et al. Partial Rescue of F508del-CFTR Stability and Trafficking Defects by Double Corrector Treatment. Int J Mol Sci. 2021;22. doi:10.3390/ijms22105262
- Hopkins C, Onweni C, Zambito V, Fairweather D, McCormick K, Ebihara H, et al. Platforms for Personalized Polytherapeutics Discovery in COVID-19. J Mol Biol. 2021;433: 166945.
- Laselva O, Ardelean MC, Bear CE. Phenotyping Rare CFTR Mutations Reveal Functional Expression Defects Restored by TRIKAFTA. J Pers Med. 2021;11. doi:10.3390/jpm11040301
- Baatallah N, Elbahnsi A, Mornon J-P, Chevalier B, Pranke I, Servel N, et al. Pharmacological chaperones improve intra-domain stability and inter-domain assembly via distinct binding sites to rescue misfolded CFTR. Cell Mol Life Sci. 2021;78: 7813–7829.
- Fiedorczuk K, Chen J. Mechanism of CFTR correction by type I folding correctors. Cell. 2022;185: 158–168.e11.
- Piovesan A, Pelleri MC, Antonaros F, Strippoli P, Caracausi M, Vitale L. On the length, weight and GC content of the human genome. BMC Res Notes. 2019;12: 106.
- Sun X, Yang Q, Xia X. An improved implementation of effective number of codons (nc). Mol Biol Evol. 2013;30: 191–196.
- Piovesan A, Vitale L, Pelleri MC, Strippoli P. Universal tight correlation of codon bias and pool of RNA codons (codonome): The genome is optimized to allow any distribution of gene expression values in the transcriptome from bacteria to humans. Genomics. 2013;101: 282–289.
- Lacy-Hulbert A, Thomas R, Li XP, Lilley CE, Coffin RS, Roes J. Interruption of coding sequences by heterologous introns can enhance the functional expression of recombinant genes. Gene Ther. 2001;8: 649–653.
- Moabbi AM, Agarwal N, El Kaderi B, Ansari A. Role for gene looping in intron-mediated enhancement of transcription. Proc Natl Acad Sci U S A. 2012;109: 8505–8510.
- Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouzé P, Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996;24: 3439–3452.
- Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991;220: 49–65.
- Zhang T, Mishra P, Hay BA, Chan D, Guo M. Valosin-containing protein (VCP/p97) inhibitors relieve Mitofusin-dependent mitochondrial defects due to VCP disease mutants. Elife. 2017;6. doi:10.7554/eLife.17834
- Blythe EE, Gates SN, Deshaies RJ, Martin A. Multisystem Proteinopathy Mutations in VCP/p97 Increase NPLOC4·UFD1L Binding and Substrate Processing. Structure. 2019;27: 1820–1829.e4.
- Johnson MA, Klickstein JA, Khanna R, Gou Y, Cure VCP Disease Research Consortium, Raman M. The Cure VCP Scientific Conference 2021: Molecular and clinical insights into neurodegeneration and myopathy linked to multisystem proteinopathy-1 (MSP-1). Neurobiol Dis. 2022;169: 105722.
- Zhang G, Li S, Cheng K-W, Chou T-F. AAA ATPases as therapeutic targets: Structure, functions, and small-molecule inhibitors. Eur J Med Chem. 2021;219: 113446.
- Evangelista T, Weihl CC, Kimonis V, Lochmüller H, VCP related diseases Consortium. 215th ENMC International Workshop VCP-related multi-system proteinopathy (IBMPFD) 13-15 November 2015, Heemskerk, The Netherlands. Neuromuscul Disord. 2016;26: 535–547.
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46: D1062–D1067.
- Niwa H, Ewens CA, Tsang C, Yeung HO, Zhang X, Freemont PS. The role of the N-domain in the ATPase activity of the mammalian AAA ATPase p97/VCP. J Biol Chem. 2012;287: 8561–8570.
- Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49: D605–D612.
- Sasagawa Y, Otani M, Higashitani N, Higashitani A, Sato K, Ogura T, et al. Caenorhabditis elegans p97 controls germline-specific sex determination by controlling the TRA-1 level in a CUL-2-dependent manner. J Cell Sci. 2009;122: 3663–3672.
- Zou C-G, Ma Y-C, Dai L-L, Zhang K-Q. Autophagy protects C. elegans against necrosis during Pseudomonas aeruginosa infection. Proc Natl Acad Sci U S A. 2014;111: 12480–12485.
- Cheung TP, Choe J-Y, Richmond JE, Kim H. BK channel density is regulated by endoplasmic reticulum associated degradation and influenced by the SKN-1A/NRF1 transcription factor. PLoS Genet. 2020;16: e1008829.
- Imamura S, Yabu T, Yamashita M. Protective role of cell division cycle 48 (CDC48) protein against neurodegeneration via ubiquitin-proteasome system dysfunction during zebrafish development. J Biol Chem. 2012;287: 23047–23056.
Seeking to make a significant impact in drug discovery? Contact us now and discover how our comprehensive drug discovery services and phenotypic screening services can expedite the development of groundbreaking therapies.


