
The Child Neurology Society honored William (Bill) Dobyns for his highly impactful efforts in characterizing child neurology. In a prolific and highly influential carrier, Bill talked about how the key influencers in his life shaped the directions he pursued. Reinforcing the title of his talk “The Names of Things, ” Bill was the first to discover and name the LIS1 gene of lissencephaly (Dobyns 1993).
What’s in a name?
A spicy little comment was made by Bill. He let it be known he did not appreciate the HUGO Nomenclature Committee’s decision to rename LIS1 to PAFAH1B1. Definitely a mouthful and a change that does not seem necessary, since LIS1 appears to have no confusion in Pubmed literature searches.
To worm it or not – Can comparative biology can help?
It appears gene humanization in the nematode may be of use in characterizing lissencephaly genes. A quasi-random sampling of 9 genes mentioned in Bill’s presentation was obtained (all the ones I wrote down in my notes!). Next, a table of properties was made:

We first ask: What is the homology to C. elegans? 8 of 9 genes are identified as homologs. All but the AUTS2 are found to have a worm gene equivalent, as scored for similarity percentage using the DIOPT website. Next, we ask Which of these homologs have sufficient similarity for application of a gene-swap technique? Using inspiration from Douglas Adams for a rather arbitrary cutoff of 42, we get 6 of 8 homologs with enough similarity that gene substitution with human cDNA has a chance of retaining function and rescuing a null phenotype. We are currently 3 for 3 in genes tried for gene-swap in epilepsy – STXBP1, KCNQ2, CACNB4 – all can function and rescue their respective C. elegans null. Finally, we ask Is there something to rescue in the Dobyns gene list? 4 of the 6 good homologs are found to be lethal (LIS1, MTOR, ZIC1 and PIK3R2), when removed by either by genetic deletion and or RNAi knock-down. For the remaining two genes (ATK2 and PC2CA), although not appearing to be lethal, they have detectable phenotypic consequence when their homolog is absent in the nematode. Bottom line, 6 of 9 have good prospects for using comparative biology to answer variant pathogenicity questions.
Symposium IV: Precision Medicine – Epilepsy, The Next Frontier
This session was of high interest to the geneticist wanting to design high-throughput animal models for use in rare disease discovery. Jeffery Loeb opened with a “Big Data” discussion on linking electrode implantation in the brain with various properties of tissue biopsies (genomics, transcriptomics, metabolomic, and histological data). It was a fascinating and bewildering look at the 1700 genes that are associated with bursting/spiking phenomena in the brain. Next was Maduri Hedge who took us on a tour of what is the latest and greatest in whole genome analysis – basically what is happening in the 5342 OMIN-identified disease genes. Much of the answer is we do not know yet, because there is an ever expanding universe of Variants of Uncertain Significance (VUS), fueled by the fact that each person averages 3,000,000 differences in sequence when compared to reference human genome. (Hedge 2017). With 1% of genome as coding, this suggest 30,000 variations occur in exonic coding sequence. The amount of this coding sequence variation that is known pathogenic is much less. Filtering out high frequency alleles and checking against databases where variant call is either pathogenic or likely-pathogenic, we get an average of 2.1 pathogenic/likely-pathogenic variants per person (Rego 2018). The same data reveals each person can expect another 17.6 variant alleles as either VUS or as unassigned in status.
Demonstration of rapid pathogenicity assessment of a VUS allele
Next presentation was a great tag-team duo of Alfred George and John Millichap speaking on the rapid pathogenicity assessment of a VUS in KCNQ2 gene. Its title, “Emerging Paradigms for Precision Medicine in Early Life Epilepsy: Bed-to-Bench Workflow” exemplified where genomic biology is now starting to hold focus. They showed data for a three week turn-around in providing functional data on a patient variant. Their data demonstrated a newly-discovered variant in KCNQ2 behaves as a loss-of-function allele. In a very short time, from genomic data to clinically-actionable results, the patient had a genetic mechanism revealed for their illness and drug treatment options became more clear. This same variant, by the way, was assessed as a VUS call, 3 months later when the genomics lab finished applying the traditional ACMG-AMP guideline assessment criteria. Obviously functional studies deployed quickly after genomic data acquisition can be quite impactful in helping find treatment choices for the clinician and their patient.
Time to Get Organized
Finally, wrapping up what proved to be a lively lecture series and a very long QnA afterward, we had Anne Berg give us a plea for coming together as a community and doing what has been done in cancer, and with Spinraza on SMA – Find ways to work together, get DNA diagnostics done as quickly as possible and then screen variations for function consequence. There is a big need for an organized effort in epilepsy. Early intervention in neurological diseases has the capacity to create a dramatic improvement in outcome. Anne provided us some data to help understand how big is the problem:
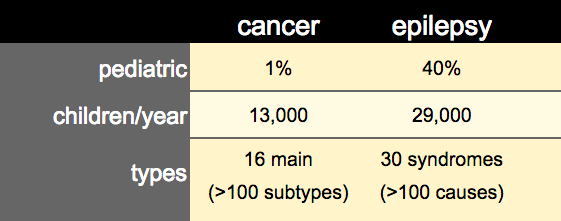
The size and coordination of effort in cancer has recently been giving us miraculous gains. For some types of cancer, what was nearly a death sentence 10-20 years ago is now, in many of today’s oncology practices, a responsive and well-controlled disease. Anne feels it is time for a profound shift in the way epilepsy is approached and treated. We need to apply the same rigor and interactivity that drove the success of cancer biology understanding. When it come to pediatric populations, cancer is less significant than epilepsy. There are 29,000 new pediatric epilepsy cases each year, while pediatric cancer cases occur at 13,000 per year – a more than 2x lower in rate. The complexity of disease types between epilepsy and cancer is of similar size, so systematically fostering the level of researcher-clinician interaction as seen in cancer biology should be highly productive for epilepsy understanding and discovery. Ultimately the epilepsy ecosystem is ripe for new approaches that can be adequately and rapidly address the complexity of epilepsy biology and uncover new therapeutic modalities.
References
Dobyns WB et al. Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA. 1993 Dec 15;270(23):2838-42. https://www.ncbi.nlm.nih.gov/pubmed/7907669
Hegde M, Santani A,Mao R,Ferreira-Gonzalez A,Weck KE and Voelkerding KV. Development and Validation of Clinical Whole-Exome and Whole-Genome Sequencing for Detection of Germline Variants in Inherited Disease. Arch Pathol Lab Med. 2017 Jun;141(6):798-805. doi: 10.5858/arpa.2016-0622-RA. Epub 2017 Mar 31. https://www.ncbi.nlm.nih.gov/pubmed/28362156
Shannon Rego, Orit Dagan-Rosenfeld, Wenyu Zhou, M. Reza Sailani, Patricia Limcaoco, Elizabeth Colbert, Monika Avina, Jessica Wheeler, Colleen Craig, Denis Salins, Hannes L. Rost, Jessilyn Dunn, Tracey McLaughlin, Lars M. Steinmetz, Jonathan A. Bernstein, Michael P. Snyder.
High Frequency Actionable Pathogenic Exome Mutations in an Average-Risk Cohort. bioRxiv 151225; doi: https://doi.org/10.1101/151225

