
The philosopher Friedrich Nietzsche once said:
“You have evolved from worm to man, but much within you is still worm.”
Genetic diversity in individuals and between species is responsible for bewildering variability and biological niche adaptation of life, yet much of the essential genes involved in disease presentation are highly conserved from yeast to humans. For instance, the direct comparison C. elegans genome to H. sapiens reveals only 44% of the genes are similar (Shaye and Greenwald 2011). Yet when one restricts the comparison to the 6460 genes known to be associated with genetic disease (1/3rd the human genome), clear similarity (orthology) to C. elegans occurs for 79% of the human disease genes (ClinVar database). This high degree of interspecies conservation between worm and human has recently become more recognized and appreciated for use in disease biology understanding (Golden 2017, Wang 2017, Wangler 2017, Apfeld and Alpers 2018)
Undiagnosed Disease
The Undiagnosed Disease Network (UDN) has been making significant strides in rare disease research and holds an emphasis on developing animal models for use as tools of variant biology discovery (Splinter 2018). Tim Schedl at Washington University in St Louis is now a recent addition to the UDN. Tim will run the C elegans nematode Model Organisms Screening Center (MOSC). The worm is proving to be a useful tool for uncovering the disease related biology (Bend 2016, Wangler 2107, Luo 2017, Chao 2017, Oláhová 2018, Liu 2018, Guiberson 2018). Perhaps the biggest place where the nematode holds promise is its proven capacity for high-thoughput screening (Leung 2013, Rangaraju 2015, O’Reilly 2016, Lucanic 2018, Partridge 2018). The Leung publication achieved an impressive 340,000 compounds tested in 1536-well format in 5 weeks. If we can find the systems that accurately model human disease in the nematode animal model, we will have developed a system ripe for high-throughput discovery of new pharmaceuticals.
Can we model clinical variant biology in the worm?
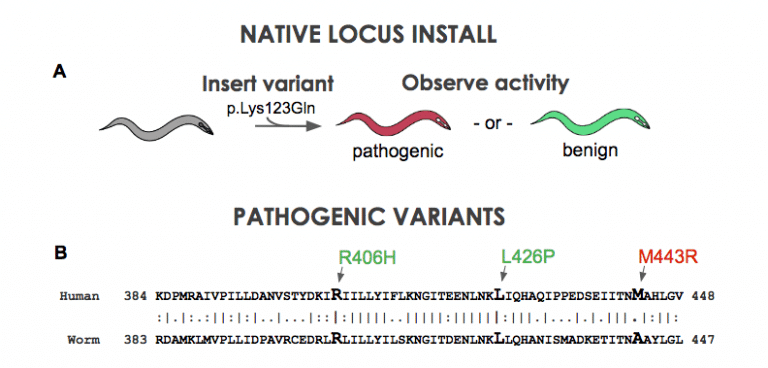
Figure 1. A) A clinical variant is installed into native homolog of the nematode. B) Position of STXBP1 pathogenic variants in the nematode unc-18 gene.
Using CRISPR and related techniques, it is now quite easy to insert DNA changes into the genome of animal models. For example, our company just hit a milestone of 2000 transgenics delivered. But the puzzle now becomes are we doing it right. What is the best transgenic configuration to address disease biology? We can put a clinical variant in the native gene of the nematode (Figure 1A). Although there is a large amount of similarity when making gene-to-gene connections in disease genes, a question arrises: is there enough similarity at the amino acid level? When one maps out the location of know pathogenic variants to their locations at the amino acid sequence, one occasionally finds that established or likely-pathogenic variants do not occur at a conserved position. For instance, in a sequence occuring in the middle of the C. elegans STXBP1 gene, three variants suspected of pathogenicity are shown (Figure 1B). The patient variants R406H and L426P are at conserved positions in the worm unc-18 homolog. Unfortunately M443R does not have the identical base in C. elegans. If we insert a Methionine (“M”) in for the worm’s Alanine will we get function? …Probably. If we put in an Arginine instead (“R”) will the gene work? …Probably not. Yet, we are left with a bit of ambiguity because location is not identical between species. A more robust way to model human biology is to put in the entire human gene into the nematode genome. To explore this concept, we have developed a technique that we call “gene-swap humanization” (Figure 2). We remove the endogenous gene of the worm and replace it with a cDNA copy of the human gene. This creates a gene-humanized animal.
Figure 2. human gene is swapped in for native sequence then observe if it can retain function.
We have only just started, but we have uncovered an interesting finding: Gene-swap humanization appears to be better than native locus for use as platform for modeling disease variant biology. We decided to compare variant installs in the native locus to installs in the humanize locus. The main driver to try this out was sequence similarity, or more precisely, the lack of it. When one compares the human to mouse for the STXBP1 gene, one see 100% identical amino acid usage between the two species. Contrast this to human sequence to the worm’s unc-18 gene – one sees only 59% identical amino acid usage. With nearly half the amino acids not being fully conserved, can we model in the native locus effectively, or….
“What will happen if we swap-in the human gene as a gene replacement of the worm gene?”
The new gene editing CRISPR technology is allowing us to address this question. Genes are now quite easy to order up as fully synthetic. The DNA sequence comes in a plasmid format that we can inject, with the right CRISPR components, directly into the gonad of the worm. The progeny of the animal will have a high propensity to have picked up the genome edit. We use PCR technology to find the edited animal and then grow up the population for testing in various activity assays (Figure 3). From concept to variant-specific animal data in less than one month.
Figure 3. comparison of native vs humanized locus. Electrophysiology of A) native vs B) humanized locus. Chemotaxis assay of C) native vs D) humanized locus.
What we found fascinating were two findings:
-
Human gene retains function in worm.
-
Variants in human gene render the worm sensitized for pathogenicity assessment.
That second observation begs more explanation. In figure 3, you will see two types of phenotyping assays performed on three known pathogenic variants of STXBP1 (R292H, R406H and R388X). When we put these patient-variants into the native locus at their sequence conserved positions, we see only the R388X gives a big deviation of function. Alternatively, when we put these variants in the gene-swapped hSTXBP1 locus, we see that all variants exhibit a statistically significant deviation of function in both assays. As a result, it appears the gene-swap humanized locus is better choice for modeling clinical variants.
“What about that first observation?” Lets not overlook that one! This shows that for this particular protein, its physiological role is highly conserved between worm and man. She/he (worms are self-fertilizing hermaphrodites – more on that later…) is giving data that suggest modeling variants in humanized worms can be useful as a platform for variant biology discovery!
Figure 4. Assay differences and similarities within and between genes. A) Electrophysiology assays on STXBP1 exhibits gain of function and loss of function phenotypes. B) Locomotion assay detects some loss of function activity. C) Chemotaxis assay see all variants as loss of function alleles. D) KCNQ2 Electrophysiology see gain of function when the kqt-1 homolog is removed.
We have found one more interesting finding in our humanized strain studies. The types of phenotyping assays used on a clinical variant will depend on the unique biology of that variant. In figure 4, we have three different activity assays. What should jump out at you is that each assay has a unique response. The R292H and R406H variants have gain-of-function response in the electrophysiology, while the R388X has lost function. In the next assay, the animal locomotion measurement, we see wild-type activity in R292H, while R406H and R388X are exhibiting movement deficiencies. In the last highly-sensitive chemotaxis assay, only R292H retains an ability to sniff out and go to food – the remaining two, R406H and R388X, are not capable of getting to food in the 1 hr test period of the assay. One last observation for this figure – there is high similarity in electrophysiology response for the KCNQ2 knock-out and the R292H and R406H variants in STXBP1. All three (KCNQ2 knock-out and the two STXBP1 variants) show gain-of-function response. This is expected if all these variants lead to M-current modulation of potassium channels. What we speculate based on prior findings (Devaux 2016) is the R292H and R406H are defective in chaperone function of syntaxin. They no longer prevent syntaxin from being an inhibitor of potassium channels and effectively these STXBP1 variants have a physiological mode of action leading to sodium channel inhibition. Prediction: ezogabine will be useful on patients with hSTXBP1(p.R292H) and hSTXBP1(p.R406H) as a treatment for reversing their levels and intensity of epilepsy seizures, but probably be contraindicated for hSTXBP1(p.R388X) clinical variants.
Biosensors
Figure 5. Genetically-encoded biosensors of transcriptional activity induced by patient variant.
The last component of this blog post is to discuss formats for making the animals perform drug screening in high-throughput formats. Loss of locomotion can be a powerful screen, but what might be more sensitive is the use of genetically-encoded biosensors. The biosensor formats we favor most are the transcriptionally-activation reporter systems (Figure 3). Typically these are GFP/RFP combos where one reporter is driven by a promoter that is either up regulated or down regulated upon being in a particular genetic background. Loss-of-function alleles can have a myriad of gene transcriptional effects and biosensor reporters have previously been used to dissect physiological pathway interactions in C. elegans (Urano 2002, Lea 2005, Kahn 2008).
Figure 6. Two biosensors
We have used biosensors to probe drug response in nuclear hormone receptor signaling (Figure 6). We compared biosensor responses to assays of behavior. Perhaps what is most striking about the data is the sensitivity. In the dafadine assay we see nearly 10x higher sensitivity with the biosensor based assay. From a drug discovery standpoint, this means 10x less compound is need in order to see an effect. For the dafachronric acid assay, the result is even higher – 30x higher than the biological activity output. For those who do drug discovery, the application of biosensors to clinical-variant profiling will be worth the investment in biosensor construction – cost saving will be obtained from reduced consumption of the expensive compounds used in a drug library.
References
Apfeld J et al. What Can We Learn About Human Disease from the Nematode C. elegans? Methods Mol Biol. 2018;1706:53-75. doi: 10.1007/978-1-4939-7471-9_4. https://www.ncbi.nlm.nih.gov/pubmed/29423793
Bend E et al. NALCN channelopathies: Distinguishing gain-of-function and loss-of-function mutations. Neurology. 2016 Sep 13;87(11):1131-9. doi: 10.1212/WNL.0000000000003095. https://www.ncbi.nlm.nih.gov/pubmed/27558372
Devaux, J. et al. A possible link between KCNQ2- and STXBP1-related encephalopathies: STXBP1 reduces the inhibitory impact of syntaxin-1A on M current. Epilepsia. 2017 Dec;58(12):2073-2084. doi: 10.1111/epi.13927. https://www.ncbi.nlm.nih.gov/pubmed/29067685
Golden A. From phenologs to silent suppressors: Identifying potential therapeutic targets for human disease. Mol Reprod Dev. 2017 Nov;84(11):1118-1132. doi: 10.1002/mrd.22880. Epub 2017 Oct 3. https://www.ncbi.nlm.nih.gov/pubmed/28834577
Kahn NW et al. Proteasomal dysfunction activates the transcription factor SKN-1 and produces a selective oxidative-stress response in Caenorhabditis elegans. Biochem J. 2008 Jan 1;409(1):205-13. https://www.ncbi.nlm.nih.gov/pubmed/17714076
Leung CK et al. An ultra high-throughput, whole-animal screen for small molecule modulators of a specific genetic pathway in Caenorhabditis elegans. PLoS One. 2013 Apr 29;8(4):e62166. doi: 10.1371/journal.pone.0062166. Print 2013. https://www.ncbi.nlm.nih.gov/pubmed/23637990
Liu, N. et al. Functional variants in TBX2 are associated with a syndromic cardiovascular and skeletal developmental disorder. Hum Mol Genet. 2018 Jul 15;27(14):2454-2465. doi: 10.1093/hmg/ddy146. https://www.ncbi.nlm.nih.gov/pubmed/29726930
Lucanic M et al. A Simple Method for High Throughput Chemical Screening in Caenorhabditis Elegans. Journal of visualized experiments : J Vis Exp. 2018 Mar 20;(133). doi: 10.3791/56892. https://www.ncbi.nlm.nih.gov/pubmed/29630057
Luo X et al. Clinically severe CACNA1A alleles affect synaptic function and neurodegeneration differentially. PLoS Genet. 2017 Jul 24;13(7):e1006905. doi: 10.1371/journal.pgen.1006905. eCollection 2017 Jul. https://www.ncbi.nlm.nih.gov/pubmed/28742085
Oláhová, M. et al. Biallelic Mutations in ATP5F1D, which Encodes a Subunit of ATP Synthase, Cause a Metabolic Disorder. Am J Hum Genet. 2018 Mar 1;102(3):494-504. doi: 10.1016/j.ajhg.2018.01.020. https://www.ncbi.nlm.nih.gov/pubmed/29478781
O’Reilly L et al. High-Throughput, Liquid-Based Genome-Wide RNAi Screening in C. elegans. Methods Mol Biol. 2016;1470:151-62. doi: 10.1007/978-1-4939-6337-9_12. https://www.ncbi.nlm.nih.gov/pubmed/27581291
Partridge F et al. An automated high-throughput system for phenotypic screening of chemical libraries on C. elegans and parasitic nematodes. Int J Parasitol Drugs Drug Resist. 2018 Apr;8(1):8-21. doi: 10.1016/j.ijpddr.2017.11.004. https://www.ncbi.nlm.nih.gov/pubmed/29223747
Rangaraju S et al. High-throughput small-molecule screening in Caenorhabditis elegans. Methods Mol Biol. 2015;1263:139-55. doi: 10.1007/978-1-4939-2269-7_11. https://www.ncbi.nlm.nih.gov/pubmed/25618342
Rea SL et al. A stress-sensitive reporter predicts longevity in isogenic populations of Caenorhabditis elegans. Nat Genet. 2005 Aug;37(8):894-8. Epub 2005 Jul 24. https://www.ncbi.nlm.nih.gov/pubmed/16041374
Shaye DD and Greenwald I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 2011;6(5):e20085. doi: 10.1371/journal.pone.0020085. Epub 2011 May 25. https://www.ncbi.nlm.nih.gov/pubmed/21647448
Splinter K et al. Effect of Genetic Diagnosis on Patients with Previously Undiagnosed Disease. N Engl J Med. 2018 Oct 10. doi: 10.1056/NEJMoa1714458. https://www.ncbi.nlm.nih.gov/pubmed/30304647
Urano F et al. A survival pathway for Caenorhabditis elegans with a blocked unfolded protein response. J Cell Biol. 2002 Aug 19;158(4):639-46. Epub 2002 Aug 19. https://www.ncbi.nlm.nih.gov/pubmed/12186849
Wang J et al. MARRVEL: Integration of Human and Model Organism Genetic Resources to Facilitate Functional Annotation of the Human Genome. Am J Hum Genet. 2017 Jun 1;100(6):843-853. doi: 10.1016/j.ajhg.2017.04.010. Epub 2017 May 11. https://www.ncbi.nlm.nih.gov/pubmed/28502612
Wangler, M. F. et al. Model Organisms Facilitate Rare Disease Diagnosis and Therapeutic Research. Genetics. 2017 Sep;207(1):9-27. doi: 10.1534/genetics.117.203067. https://www.ncbi.nlm.nih.gov/pubmed/28874452
Zhou, P. et al. Novel mutations and phenotypes of epilepsy-associated genes in epileptic encephalopathies. Genes Brain Behav. 2018 Jan 4. doi: 10.1111/gbb.12456. https://www.ncbi.nlm.nih.gov/pubmed/29314583

