
When you receive your genomic report, you have a movement of trepidation.What will it say? Will it have something that says you should do countermeasures immediately? Will it say something that you can do nothing about? The latter condition occurred for me. There were findings that had a strong impact on my psyche.
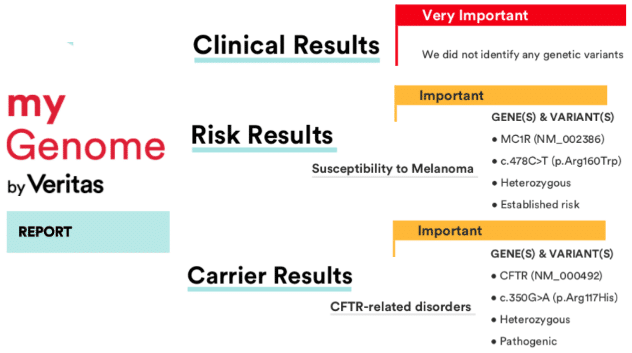
Two things were called out heavy. The first was a cancer risk of melanoma (MC1R(R160W) locus). This did not concern me too greatly, as first my mom, and now my spouse have been diligent in their liberal application of sunscreen to the family. Once I googled and pubmed searched the MC1R(R160W) locus, I found the evidence was less than compelling for a dramatic change of lifestyle. Just keep the sunscreen coming and I will likely be fine.
The carrier result was a little more of a shocker. A good personal friend has a daughter homozygous in this gene. It was discovered in utero and they have been vigilant ever since. Their daughter is now in her teens. Doing exceptionally well and acting like any normal kid – currently enthralled with dance class and other outdoor activities. She is a great example of preventative medicine done right. So getting tagged as pathogenic in this gene is giving me mixed feelings. A mix of some worry and yet, almost pride. Even though my good friends don’t share my specific genetic lesion, it still feels very personal and connecting. Furthermore, this is one of the genes where modern genomic medicine is making great progress in understanding and treatment.
Will you also be a carrier of a pathogenic variation?
Carrier status is something all of us should expect. Veritas recently publicly disclosed at the Precision Medicine World Congress that their database has 90% of customer reports as returning with carrier status for at least one pathogenic variant. Recent discussions with Robert Green at Harvard confirm this – he showed me a large dataset that gave the number as 92% of healthy populations as being carriers for known pathogenic variants. You might think that there are a lucky few (10%) who are not carriers, but think again. The average person will have close to 3 million differences from the reference genome and this may be an underestimate. Distribute that unbiased across the genome and we have coding regions with close to 50 thousand variations since you have roughly 20 thousand genes. that means every gene has a approximately 2 variations in it. Now lots of approximating, and does not factor in selection against bad variations. Yet in that quick calculation, the main message is every gene is likely to have a variation and some genes will have multiple variations. So the original question of how many of these are pathogenic, becomes difficult to approximate. Publications suggest we may have up about 1300 suspect variations hiding in our genome. Yet definitive variants with “known” pathogenicity is likely to be much lower in your genome.
Complicating this is issue is variable penetrance – a pathogenic variant in one family may behave with monogenic behavior in that family. While in another family, that same variation may be acting more polygenic – it needs other gene mutations to have pathos in the patient. It is behaving more like a “risk factor” for disease.
Pathogenic variant frequency in Chris Hopkins’ genome
The vagueness of my carrier status “kills” me. I wanted to know more, so I contacted a good friend at the Rady Children’s Hospital, Dr. Matthew Bainbridge. Dr. Bainbridge is a researcher who was a key contributor to the Rady’s renowned speed at using whole genome sequencing for rapid genetic diagnosis. Dr. Bainbridge introduced me to some software tools he has been developing. His company Codified Genomics has developed a variant analysis software that allows exploration of one’s genomic variants. All you need is your BAM or VCF files.
If you are unsure what a BAM or VCF file is, don’t worry, let’s decode the jargon. In the clinphen journey to understand my clinical predilections, predispositions, and pathos, I found myself getting immersed into the intricacy of the end-to-end solution in genomic data acquisition and interpretation. What happens when you spit in a tube and put it in the mail? A lot of stuff! I came across an amazing guide to understanding the industry space behind genomic sequencing, the Enlightenbio Report. This guide helped me get a tightly-focused view on the process of understanding one’s DNA.
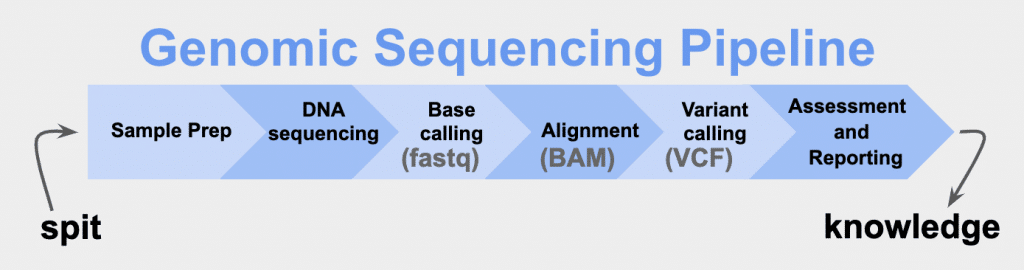
That first box is what happens after you spit in the tube. The chemicals in the tube react with the cellular material in the spit to help stabilize it and prevent its degradation. This allows one to send the sample at room temperature to the lab. Upon receiving the tube, the lab initiates a protocol to isolate the DNA that comes from the mouth’s epidermal cells that slough off into your spit. The DNA is manipulated in such a way that it can go onto a microchip slide and a set of DNA sequencing chemistry reactions are used to read out the DNA in small segments of sequence. Each of the millions of sequence segment reads is recorded as a fastq file. The fastq read segments are compared and aligned to a reference genome to make a BAM file. The BAM file alignments are processed to detect where sequence variation occurs, which is recorded as a VCF file. VCF files are analyzed by comparison to databases and assessments are made of each variant’s potential for pathogenicity. The assessment data is generally provided as a report to the clinician (or the intrepid genome wanderer such as myself). This report takes the raw data and massages it into a format for easier understanding of what is the baggage of one’s genome.
1604 suspect variations in my genome
Dr. Bainbridge helped me upload my VCF files into the Codified program. Next, he showed me how to sift through the data by various aspects such as allele frequency, dominant and recessive status, known pathogenic genes, etc.. The upload to Codified indicates I have exactly 1604 rare variations occurring at an appreciable fraction of the reads, and at positions inside, or in close proximity to, the coding sequence of my genes. These variants are suspect because they may alter protein function or levels of expression for the identified genes. If we just limit the dataset to changes that alter amino acid composition (nonsynomous), we get 875 gene variations. Add back potential splicing issues, indels, and aberrant start and stop codon issues, we are back up to 1440 variants as variations that are highly suspect for effecting gene expression and function.
316 MIM variant hits in my genome
What happens if we limit the entire 1604 to only those genes with recognized involvement in disease? We get 316 variants occurring in genes as recognized by the Mendelian-inheritance-in-Man (MIM) database for being disease-associated genes. When we restrict this set to coding issues only, we get 281 suspect variants.
I get a clean bill of health when I get a physical exam, so can I disregard these 281 suspect variants?
One easy step is to filter for carrier only status. 111 variants are clearly identifiable as only autosomal recessive (AR). I would require two hits in each of the paired chromosome copies to have these be of concern. Since no paired hits were detected, we can dismiss these genes as in need of my immediate concern. As a result, we are now only concerned about hits in genes with known autosomal dominant (AD) issues. These are the genes where only one bad hit is needed to render them pathogenic. Bottomline, 170 gene variants in my genome are worthy of further contemplation.
How frequent is frequent in my 170 gene variants?
There is good rational that one should only be concerned about a hit in a gene with AD propensity if it is rare in the population. The thinking is that if a variation is deleterious by itself (AD), it cannot be tolerated at a high level in the human population. Contrast this to the recessive (AR) variants (also called “alleles” when talking about frequency). My known AR pathogenic variant in the CFTR gene is in the human population at 0.0014 minor allele frequency. This high allelic frequency is tolerated in the human population because you need two hits in each gene copy in order to have a syndromic issue. Autosomal dominant alleles must have much lower frequency. If we cull the 170 for variations that occur at 0.00001 minor allele frequency (MAF) or lower, we get 53 gene-codon-altering variations to be concerned about. Examining the list manually gave me 17 genes for which I hold varying degrees of concern, of which I list the top 10:
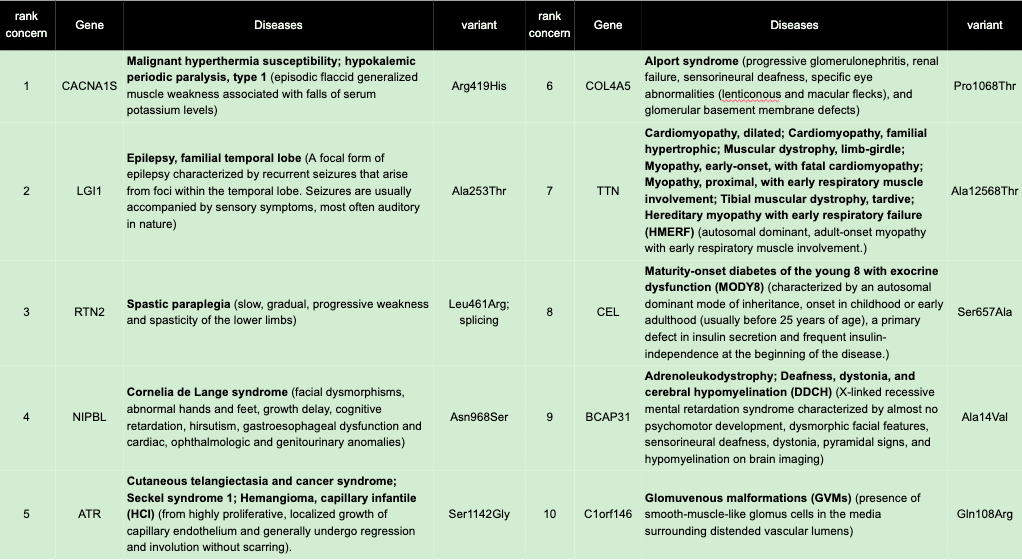
None are in the ACMG59
In a prior blog post, I described the list of genes that can be included in a clinical report as secondary findings. These are allowed in a report because the 59 genes have known actions that can be taken to mitigate their negative health effects. None of my genes of concern are in this group, so the immediate actionability is absent for my findings about the baggage in my genome. In fact, the genes I am listing as genes I am concerned about actually do not significantly bother me that much. I am still alive and in good health. If I had had pathogenic variations in these genes, the negative health consequence would have manifested many years ago. Yet, the three which I hold highest concern for are CACNA1S, LGI1, and RTN2.

The variation in CACNA1S (p.R419H) may sound like a benign and conservative change in amino acid composition, but it occurs in a highly-conserved region – it is present as an Arginine (“R”) in humans, mice, fish, flies and worms. This invariant use of R implies protein function will be compromised when the position is substituted with a histidine. The LGI1 (p.A253T) variant is also a conserved amino acid change, but it is in a less conserved region which might tolerate an Arginine to Threonine change. The RTN2 is a complex variant as it does two significantly alarming changes. The first is that it makes a dramatic Leucine to Arginine change in the 4th exon up from the end of the protein. The second change it makes also occurs immediately adjacent to the splice junction acceptor site, which possibly leads to a loss of proper splicing in a highly conserved region of the protein.
It is likely that all three of these genes are messed up for their function. But what is not clear is the type of mess-up. Are they leading to loss of function (LOF)? Or do they lead to dominant gain of function (GOF)? These variations are most likely of the the LOF category. Otherwise, I would almost certainly be dealing with the disease symptoms that the GOF variant’s manifest. Yet this is just supposition – a hypothesis. We don’t yet have solid evidence for what is going on.
How can we get final answer for whether or not these variations are pathogenic?
To get precision answers, we could model all of these variants in C elegans. For the CACNA1S and the RTN2, their high conservation from human to worm would allow direct modeling in the worm’s homologous position of the worm’s native gene (“Native locus”).
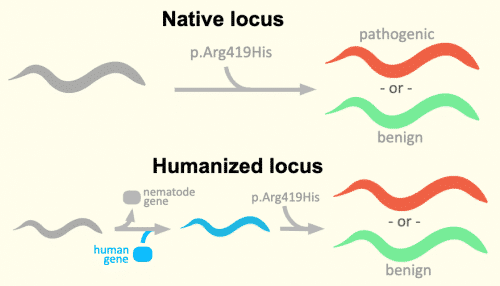
Our prior work with full gene humanization indicates more congruent results occur if we first swap in a human gene for the native gene locus then install a variant (“Humanized Locus”). The use of a humanized locus allows modeling of any variant, whether it is highly conserved or not across many species. So far, all known pathogenic variants behave with deviant behavior when put into humanized systems. Contrast this to insertion in a native locus, where some known pathogenic alleles did not create any detectable deviance of behavior.
For the three genes to which I am concerned, all are of a favorable size such that a human sequence can be easily optimized and installed for expression from the worm’s native locus (“Humanized” animal). If we observe the human gene can rescue loss-of-function, we will know we are off to the races and can study variant biology in gene-humanized systems. The humanized animals will be precision proxies serving as clinical avatars of the patient condition. CACNA1S is a drugable target. The creation of a humanized system expressing CACNA1S as a gene replacement of egl-19 gene would generate a platform for drug discovery of which patient variants might be responsive to the calcium channel blockers of benzothiazepines, phenylalkylamines and 1,4-dihydropyridines. The end result is highly-personalized medicine approaches that identify drug treatments specific to the patient’s genetic pre-conditions.

