
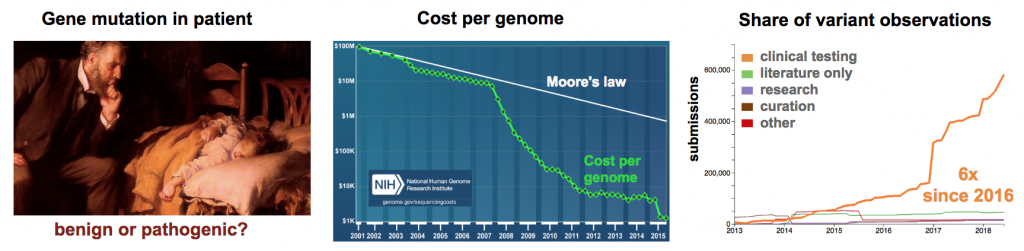
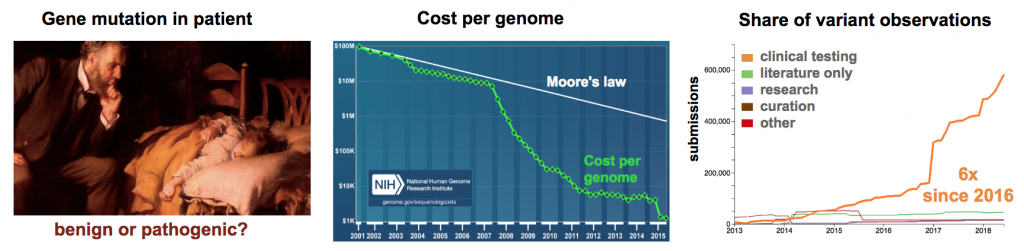
Genetic Testing
Clinical geneticists have an acute need to understand pathogenicity in genomes of their patients (Figure 1). Cost per human genome has now approached $1000 each (Wetterstrand 2018). This affordable cost is allowing clinicians to start incorporating next-generation sequencing (NGS) technology into the patient diagnosis.
Variant Diversity
The American College of Medical Genetics and Genomics and the Association for Molecular Pathology recommend that variants be classified in five groups (Pathogenic, Likely Pathogenic, Uncertain Significance, Likely Benign, Benign) (Richards 2015). Further, they suggest greater efforts are needed to resolve variants of Uncertain Significance (VUS) into either Pathogenic or Benign status. The re-classification of a VUS is no small task. In a recent publication on the analysis of the clinical variant database (ClinVar), the number of known genetic sequence variants in human disease was reported on 9/9/2016 to be 72,472 (Manolio 2017). One year later (9/21/2017), a survey of ClinVar using an online database viewer reported 359,938 known variants (Henrie 2018). This 4.9-fold increase in the number of known clinical variants over one year reflects the explosive application of whole genome sequencing in clinical diagnostics (Stavropoulos 2015, Ellingford 2016, Volk 2017).
Need for Innovation
The daunting task now is to determine the significance of each new variant. Bioinformatics can provide some insight into variant pathogenicity (Oliver 2015). Unfortunately, the number of VUS alleles has remained close to 40% year-on-year since 2015. As of the July 2018, there are 192,843 identified VUS alleles. With such high numbers, there is a pressing need to quickly correlate genotype to phenotype and determine if VUS alleles are benign or pathogenic (Cox 2015). Model systems that reconstitute mutations in a physiological context are a robust method to demonstrate variant pathogenicity (Eilbeck 2017). Traditionally, mouse models have been used to characterize defective function in VUS alleles, but the expanding universe of clinical variants is overwhelming the current capacity. Higher throughput animal modeling is needed to address the growing demand.
1. Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP) Accessed July 12, 2018. https://www.genome.gov/sequencingcostsdata
2. Richards S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015 May;17(5):405-24. doi: 10.1038/gim.2015.30. Epub 2015 Mar 5. https://www.ncbi.nlm.nih.gov/pubmed/25741868
3. Manolio TA et al. Bedside Back to Bench: Building Bridges between Basic and Clinical Genomic Research. Cell. 2017 Mar 23;169(1):6-12. doi: 10.1016/j.cell.2017.03.005. https://www.ncbi.nlm.nih.gov/pubmed/28340351
4. Henrie A. et al. ClinVar Miner: Demonstrating utility of a Web-based tool for viewing and filtering ClinVar data. Hum Mutat. 2018 May 23. doi: 10.1002/humu.23555. https://www.ncbi.nlm.nih.gov/pubmed/29790234
5. Stavropoulos DJ et al. Whole Genome Sequencing Expands Diagnostic Utility and Improves Clinical Management in Pediatric Medicine. NPJ Genom Med. 2016 Jan 13;1. pii: 15012. doi: 10.1038/npjgenmed.2015.12. https://www.ncbi.nlm.nih.gov/pubmed/28567303
6. Ellingford JM et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016 May;123(5):1143-50. doi: 10.1016/j.ophtha.2016.01.009. https://www.ncbi.nlm.nih.gov/pubmed/26872967
7. Volk AE and Kubisch C. The rapid evolution of molecular genetic diagnostics in neuromuscular diseases. Curr Opin Neurol. 2017 Oct;30(5):523-528. doi: 10.1097/WCO.0000000000000478. https://www.ncbi.nlm.nih.gov/pubmed/28665809
8. Oliver GR et al. Bioinformatics for clinical next generation sequencing. Clin Chem. 2015 Jan;61(1):124-35. doi: 10.1373/clinchem.2014.224360. https://www.ncbi.nlm.nih.gov/pubmed/25451870
9. Cox TC. Utility and limitations of animal models for the functional validation of human sequence variants. Mol Genet Genomic Med. 2015 Sep;3(5):375-82. doi: 10.1002/mgg3.167. https://www.ncbi.nlm.nih.gov/pubmed/26436102
10. Eilbeck K et al. Settling the score: variant prioritization and Mendelian disease. Nat Rev Genet. 2017 Oct;18(10):599-612. doi: 10.1038/nrg.2017.52. https://www.ncbi.nlm.nih.gov/pubmed/28804138

