

Summary:
In a previous customer story we detailed our collaboration with Dr Dan Starr to generate transgenic C. elegans models for two potentially disease-causing variants in the LINC complex (LMNA and KLC4). Recently, Starr published his findings using these humanized C. elegans models. In this article we will discuss Starr’s recent publication and how he was able to leverage InVivo Bioystems’ CRISPR/Cas9 expertise.
One major utility of InVivo Biosystems’ WHAM platform is to help characterize variants of unknown significance in rare diseases. To this end, we work with researchers such as Dr Dan Starr to generate transgenic alternative animal models which can work as human avatars for diagnosing pathogenicity in rare genetic variations.
Dr Dan Starr is a professor at UC Davis, and is one of the heads of the joint Starr/Luxton laboratory which is interested in understanding the mechanism and function of the physical coupling between the cell nucleus and cytoskeleton. In his most recent publication, Starr collaborated with InVivo Biosystems to generate humanized C. elegans models of Hereditary Spastic Paraplegia (HSP), a rare disorder which is characterized by the progressive weakening and stiffness of the leg muscles (NHS, 2022). Often undiagnosed, it is difficult to know exactly how prevalent this disorder is – estimates range from 1 in 11,000 to 1 in 77,000.
It is suspected that clinical variants in the kinesin light chain gene, KLC4, may cause HSP. The C. elegans nematode was engineered so that the worm’s gene klc-2 was replaced with the human version, KLC4 [Figure 1].
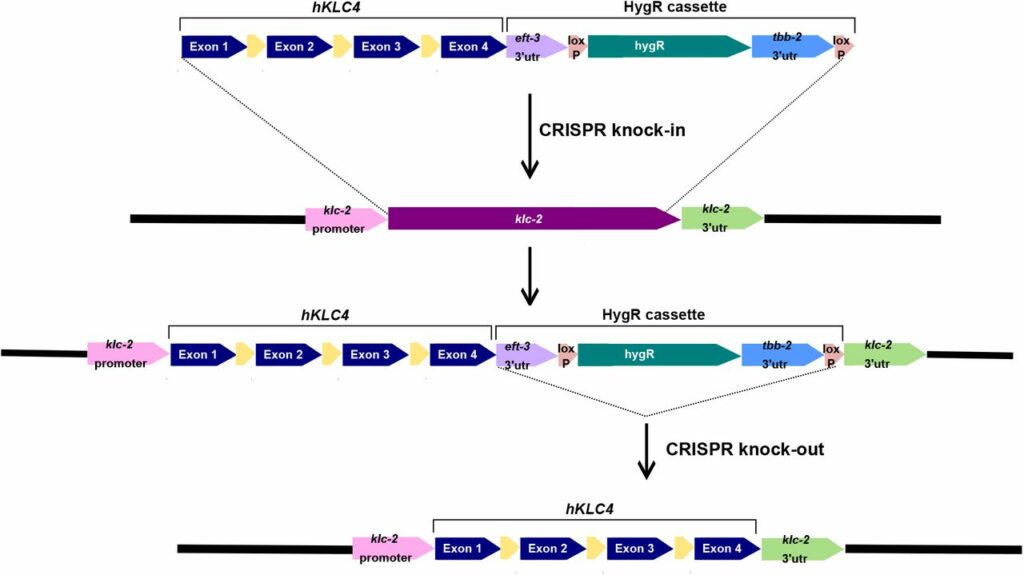
Figure 1: The CRISPR/Cas9-mediated genome editing workflow used to generate hKLC4 worms. Inserted sequence contains human gene KLC4 (exons shown in blue; synthetic introns shown in yellow) and hygromycin gene resistant selection cassette (HygR).
After the WHAM hKLC4 strain was successfully generated, their motility fitness was compared with the wild type, and it was found that while the humanized line had a defect in movement (body bending). The animal shows a partial retention of swimming activity, indicating that a swimming assay was a suitably sensitive enough to assess the effects of potentially pathogenic variants [Image 1].
Next, four missense variants were introduced into the hKLC4 animals (R72H and R358H (predicted to be begin), and A295P and T381I (predicted to be pathogenic). Lastly, the clinical variant of unknown significance: G369fs was introduced.
Of the four missense variants, only the T381I variant was strongly expected to disrupt KLC4 function human cells. When tested, the T381I variant exhibited a severe locomotion defect (Image 1). Intriguingly, when the clinical G369fs variant was examined, an even more severe effect of early lethality was observed. To more accurately model the autosomal dominant nature of the human disease, the heterozygote of G369fs was examined an a defect in nuclear migration was observed, which may correlate with late onset Hereditary Spastic Paraplegia. The authors found the findings encouraging but stressed that more research is needed in order to determine if a loss-of-function KLC4 variant can cause this disorder.
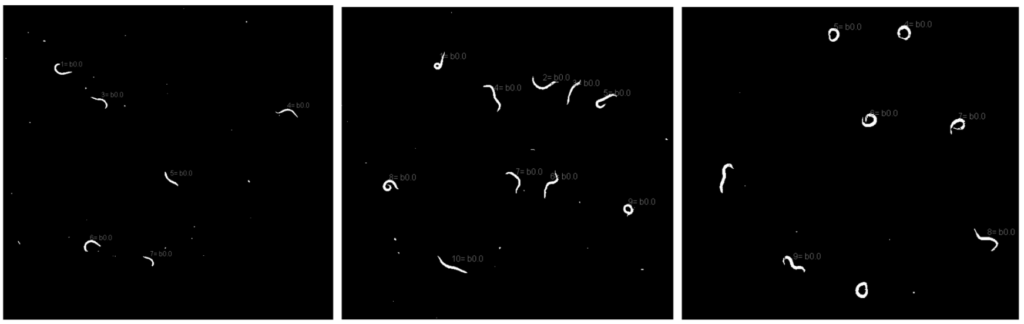
Image 1. Screenshot examples of worm motility videos.
Wildtype (N2) worms show no motility defects (left). hKLC4 worms have a significant motility defects, but they retain most of their swimming ability (middle). While hKLC4 T381I worms have a severe motility defect (right).
Future Implications:
The success of this work highlights the ease of working with an animal model such as C. elegans. This model enables a speedy and cost effective alternative to the more traditional mammalian methods – these are notable benefits for any research project, but particularly when working in the rare disease research space, where time and funding is often limited. The ability for this genetically engineered model to test the physiological impact of human KLC4 variants is encouraging as it opens up the potential for this platform to be utilized in other, similar, neuromuscular diseases.
While specific rare diseases only impacts a fraction of the world’s population, there are thousands of rare diseases: for instance, a wide range of human diseases have been shown to be caused by mutations within the LINC complex alone (Meinke, P., Nguyen, T. D., & Wehnert, 2011). Thus, this humanized platform can lead to insights in the function of KLC4, and the progression of neuromuscular diseases such as Hereditary Spastic Paraplegia.
Read more about rare disease research:
- The path to affordable therapeutics in rare disease – Tackling congenital disorder of glycosylation in the PMM2 gene.
- A Gene Replacement Humanization Platform for Rapid Functional Testing of Clinical Variants in Epilepsy-associated STXBP1.
- From Unknown To Known: How The Zebrafish Model Is Playing A Vital Role In The Undiagnosed Diseases Network
References:
- S Gumusderelioglu, L Resch, T Brock, GWG Luxton, QKG Tan, Hopkins, C. Starr, D. (2023). A humanized Caenorhabditis elegans model of Hereditary Spastic Paraplegia-associated variants in kinesin light chain KLC4. bioRxiv; doi: https://doi.org/10.1101/2023.01.07.523106
- NHS (2022). Hereditary spastic paraplegia, NHS; https://www.nhs.uk/conditions/hereditary-spastic-paraplegia/
- Meinke, P., Nguyen, T. D., & Wehnert, M. S. (2011). The LINC complex and human disease. Biochemical Society transactions, 39(6), 1693–1697. https://doi.org/10.1042/BST20110658

